No Magic Bullet for RAS
, by Kevin Haigis

Dr. Kevin Haigis
RAS mutation is among the most significant issues facing oncologists today. As highlighted in Channing Der’s essay from September 22, 2014, RAS is the most frequently mutated oncogene family (with KRAS being the most frequently mutated oncogene) in human cancer. But the frequency of RAS mutation is not the heart of the problem. The problem facing oncologists is the effect that RAS mutation has on the response of a cancer to therapy and on the overall prognosis of patients who develop cancer expressing mutationally activated RAS. The most glaring illustration of the RAS problem concerns its effect on the response to inhibition of the epidermal growth factor receptor (EGFR). In both lung and colorectal cancer (CRC), mutation of KRAS (as well as NRAS in CRC) is associated with lack of response to drugs targeting EGFR. Indeed, the strong association between mutant RAS and resistance led the American Society for Clinical Oncology (ASCO) to recommend that all cancer patients undergo RAS mutation testing prior to receiving anti-EGFR therapies1. Because of examples like this, and following the successes of directly inhibiting activated oncoproteins in other contexts (especially kinases in non-small cell lung cancer), it stands to reason that a direct GTP-competitive inhibitor that locks RAS into its inactive conformation would make an unprecedented impact on the field of medical oncology. While RAS is commonly referred to as “un-druggable,” there has been some recent success on this front (see Ken Westover’s essay from April 29, 2015). Nevertheless, it remains unclear whether a direct RAS inhibitor is anything more than a pipe dream. Note that even if a direct inhibitor of RAS did exist, there is no guarantee that it would be universally effective. Vemurafenib, a BRAF inhibitor, is very effective against melanomas expressing BRAFV600E, but not against CRCs with the same mutation2. This is, perhaps, the best example of how context determines therapeutic efficacy, a concept that is discussed in more detail below.
In his 2015 State of the Union address, President Obama introduced the Precision Medicine Initiative (PMI) designed to “revolutionize how we improve health and treat disease”. The announcement was followed by a Perspective in the New England Journal of Medicine by the directors of the NIH and NCI with a more scientific description of the PMI3, which will have a major focus on personalized medicine for cancer. Cancer really is the ideal context in which to apply personalized medicine because somatic genetic analysis can identify driver mutations within an individual’s cancer that, in some cases, reveal opportunities for targeted therapies. Again, this is most effectively demonstrated by the successes in targeting oncogenic kinases with small molecule inhibitors. There is no doubt that RAS will be at the center of the PMI as it matures and evolves. In particular, I believe that RAS will play a major role in how we define the term “precision.” How precise is precise enough when it comes to RAS?
Precision medicine for RAS (again, in the absence of a direct inhibitor) requires that we understand in great detail the oncogenic properties of the mutant oncoprotein. But where will we find answers to the complex question of RAS oncogenicity? The RAS field has gone through a fascinating evolution over the past four decades. Like many oncogenes, the study of RAS originated in the field of virology, with a major focus on characterizing its biochemical activity and regulation. In the early days, it was commonplace for papers to discriminate between the viral and cellular forms of RAS (v-RAS and c-RAS), the distinction being that the viral form was transforming and the cellular form was not. Once RAS mutations were discovered in human cancers4-6, the field began to focus on the cell biology of RAS oncoproteins, discerning how RAS is processed and regulated, and how it signals to downstream effector pathways. Although it was known from the early days of studying RAS that there are three cellular homologs of the two viral oncogenes, most early studies utilized the activated form of HRAS that was isolated from EJ bladder cancer cells, and this was typically referred to in published studies as RAS or RASV12. Only in the past decade has it become the norm to discriminate between the different cellular isoforms (see below). It is during these past ten years that the RAS field has evolved into a therapeutics field, with the major goal of translating the knowledge gleaned from cell biological studies into therapeutic opportunities. There are several essays on this website that discuss different ways to target RAS and RAS signaling in cancer, but, as of yet, none of these approaches has demonstrated any clinical effectiveness.
Owing to the recent advances in DNA sequencing and the large-scale efforts to sequence cancer genomes, the field of cancer genetics is in the midst of a renaissance. Without a doubt, this is the perfect time for the PMI, with its focus on cancer, to commence. Given that there are no viable therapeutic strategies for RAS, now might also be the time to take a step back and ask what we can learn about RAS – and what therapeutic insights can be gleaned – by studying the somatic genetics of cancer-associated RAS mutations. While the field seems to be searching for a magic bullet (a single therapy or therapeutic paradigm that will work for all RAS mutant cancers), it seems highly unlikely that this will exist any time soon (if ever). As an alternative, let’s explore the genetic evidence and supporting studies that lead to the conclusion that, in the absence of a direct inhibitor, targeting RAS may require a variety of therapies – a RAS first aid kit – that are employed under different genetic and physiological contexts.
Isoform Differences
While RAS is indisputably the most commonly mutated oncogene family, the frequency of mutation in each RAS family member depends upon where one looks. Again, Channing’s prior essay does an excellent job of summarizing the phenomenon of mutational selectivity. What is the molecular mechanism underlying this differential mutation frequency? Is it telling us something about the function of RAS proteins? There is no doubt that the four members of the RAS protein family behave much the same in biochemical assays. Of course, pulling a RAS protein out of the cell removes all of the regulatory mechanisms that the cell uses to create functional specificity. To clearly understand whether RAS family members possess distinct functional qualities, we need to evaluate cellular and organismal studies. In some cases, the expression pattern of a RAS gene (or lack thereof) likely drives its mutation frequency. The best demonstration of this mechanism comes from Minh To and Allan Balmain and their work on chemically-induced lung tumors in mice. When mice are treated with urethane, they invariably develop lung tumors that express mutationally activated KRas7. Using a mouse in which the HRAS cDNA is knocked into the KRAS locus (this mouse had originally been generated to ask whether HRAS expressed from the KRAS promoter could rescue the embryonic lethality associated with loss of KRAS), Minh and Allan found that urethane-induced lung cancers could now develop with activating mutations in HRAS8. While there are definitely caveats to this experiment – for example, how does the over-expression of HRAS in the knock-in affect its localization and therefore its signaling function? – it provides a nice example of how the endogenous expression pattern of a gene may influence its mutational pattern in cancer. This might be a general phenomenon for HRAS, which is far and away the least commonly mutated family member across all human cancers.
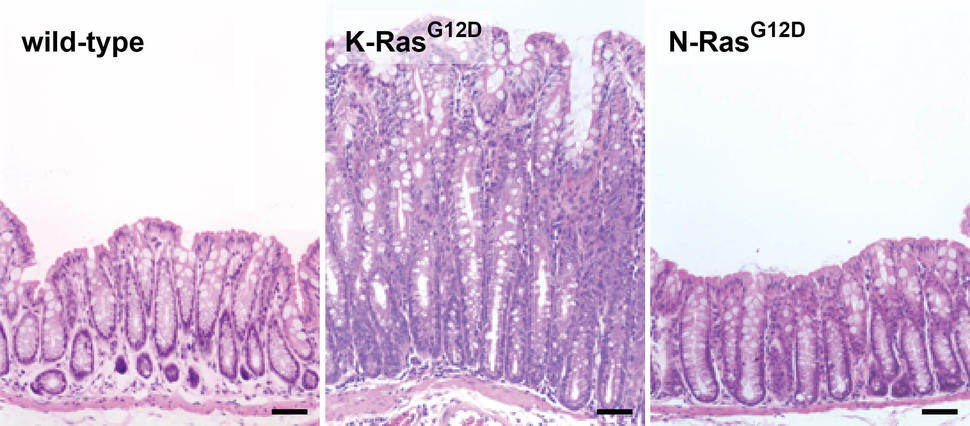
Figure 1. Effect of RAS mutations on colonic homeostasis in mice. Expression of KRASG12D induces benign hyperplasia, which never progresses to cancer, but expression of NRASG12D has no effect on basal homeostasis. Adapted from Ref. 14.
I don’t believe that expression patterns underlie all of the differences in mutation frequency between RAS family members, however, and in particular the differences between KRAS and NRAS. More likely, mutational selectivity arises from functional differences. What is the experimental evidence that RAS family members are actually functionally distinct from one another? While ectopic expression of RAS family members commonly produces similar effects in low-resolution assays (like transformation of fibroblasts), higher resolution cellular assays, for example differentiation of endoderm from stem cells9, detect clear differences. Several years ago, to explore potential isoform specific functions in an organismal setting, we genetically engineered mice to express mutationally activated KRAS, NRAS, or HRAS from their endogenous loci specifically in the intestinal epithelium. This effort was founded upon the genetic data from human colorectal cancer (CRC), where KRAS mutations are much more common than NRAS mutations (41% vs. 3%) and HRAS mutations have never been found10-13. KRAS mutations and NRAS mutations are mutually exclusive and the most obvious explanation, from a genetic point of view, is that mutual exclusivity = redundancy. For me, this equation didn’t add up. If two mutations are functionally redundant, I would expect them to occur at equal frequency. (Note that in CRC, several different genes in the TGF-beta pathway can be mutated and they are all mutated at roughly the same frequency.) We found that endogenous HRAS RNA is not detectable in the mouse colonic epithelium, but KRAS and NRAS appear to be expressed at similar levels, although single cell resolution of expression has yet to be achieved. Expression of KRASG12D leads to chronic hyperplasia due to hyperproliferation of progenitor cells in the colonic epithelium, but NRASG12D does not affect basal homeostasis (Fig. 1)14. Instead, NRASG12D inhibits the ability of intestinal epithelial cells to undergo apoptosis in response to stress, promoting CRC specifically in the context of chronic inflammation13. We traced this distinct NRAS function to its ability to activate both canonical (MAPK) and non-canonical (STAT3) effector pathways. Based on these studies, our interpretation of the mutual exclusivity of KRAS and NRAS mutations in human CRC is that their distinct oncogenic properties are positively selected under different environmental conditions.
Since this initial demonstration, additional studies have revealed biological differences between isoforms using genetically engineered mice. Ben Braun, Qing Li, and Kevin Shannon demonstrated that KRASG12D and NRASG12D are quantitatively different in their ability to induce myeloproliferative disease, but are qualitatively different in their ability to promote leukemia; KRASG12D largely promotes T-cell acute lymphoblastic leukemia (T-ALL), while MPD expressing NRASG12D progresses to acute myeloid leukemia (AML)15,16. More recently, Hugh Bender and David Gutmann used our mice to establish a role for activated KRAS in neural stem cell homeostasis, while activated NRAS and HRAS did not play a role17. As more studies are done, I am sure that the field will continue to identify isoform-specific functions relevant to the biology of different organs, and this information will help us understand why cancers select for mutation of one RAS family member over another.
If each RAS isoform has a distinct biological function, it makes sense that each isoform is likely to have a specific therapeutic approach. For example, in the context of mouse CRC, we have found that tumors expressing NRASG12D are sensitive to MEK inhibition as a single agent, while those expressing KRASG12D are not. In a more clinical example, myelomas expressing mutant NRAS are sensitive to Bortezomib (a proteasome inhibitor), while those expressing mutant KRAS are not18. Any functional differences between isoforms likely originate with the post-translational modifications that control subcellular localization (read the January 20, 2015 essay from Mark Philips for details). By extension, isoform-specific therapies could involve targeting RAS modifying enzymes. For example, Kevin Shannon’s lab, in collaboration with Herbert Waldmann, recently demonstrated that inhibiting the palmitoylation/de-palmitoylation cycle is an effective therapeutic approach for leukemias expressing mutant NRAS19. And we all know the story of the failure of farnesyltransferase inhibitors (FTIs) to treat cancers expressing mutant KRAS because of alternative prenylation by geranylgeranyl transferase (GGTase). But HRAS is not modified by GGTase, and so FTI therapy could be effective in cancers expressing mutant forms of HRAS. In his essay on targeting RAS membrane association, Mark makes a very interesting point about the development of therapies targeting farnesyltransferase. He says, “the lack of efficacy of FTIs could not have been predicted from the biology elucidated prior to the development of FTIs because, ironically, the discovery of alternate prenylation required the development of FTIs.” This is an excellent reminder that true precision medicine will likely require many incremental advances in our understanding of RAS biology.
Alleles
Canonical activating mutations in RAS (those identified in the earliest tumor/cell line sequencing studies, which happen to be the most common mutations across all cancer types) occur at amino acids 12, 13, and 61. It has been known for some time that different alleles exhibit variable transforming activity in cell culture assays, with G12V mutation being the strongest of the codon 12 alleles20,21, although it should be noted that G12D is the most common activating mutation in human cancers. Some of this variation may be due to differential effects on the intrinsic biochemical properties of the RAS enzyme. For example, Greg Buhrman and Carla Mattos showed that the Q61L mutant, unlike G12V, is catalytically dead in the presence of RAF1, making it the most severe type of mutation at the biochemical level22. (This is discussed in more detail in Carla’s essay from May 27, 2015.) Thus, codon 61 mutations are extremely potent and some types of cancer select for strong activating alleles. Melanoma is a prime example; the vast majority of NRAS mutations are at codon 61. Consistent with this observation from human cancers, Christin Burd and Ned Sharpless demonstrated that NRASQ61R was able to induce melanoma in mice, while NRASG12D (the allele we had generate to model CRC) was not23.
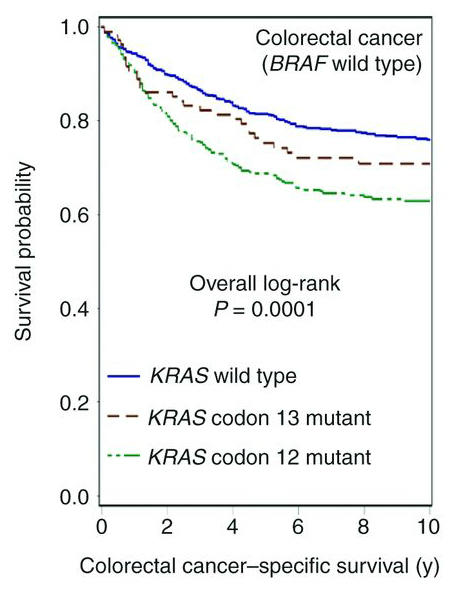
Figure 2. KRAS alleles have distinct clinical behaviors. In this example, patients with CRC expressing KRASG13Mut exhibit prolonged survival relative to patients with cancers expressing KRASG12Mut. Adapted from Ref. 24.
The current clinical viewpoint on KRAS is that all activating mutations are equivalent, so in terms of clinical management you are either mutant or wild-type. But, as with different RAS isoforms, epidemiological studies suggest allele-specific biology. For example, CRC patients with KRAS mutations at different codons exhibit significantly different clinical outcomes. While patients with codon 12 mutations exhibit reduced survival relative to patients with KRAS wild-type cancer, those with codon 13 mutations fare better (Fig. 2)24. Similarly, in lung, cancers expressing KRASG12V appear to respond better to both conventional and targeted therapies than do cancers expressing other KRAS alleles25,26. It seems that our understanding of how KRAS mutation translates into clinical behavior is not as precise as it needs to be. This lack of precision leads to patients receiving therapy to which they are unlikely to respond, but also to withholding therapy from patients likely to respond. One example relevant to this problem concerns the observation (from retrospective analyses) that patients with CRCs expressing KRASG13D appear to respond to Cetuximab, an antibody inhibitor of EGFR, while cancers with codon 12 mutations are resistant27,28. Are there other therapies currently in use clinically that might benefit patients that have specific KRAS mutations?
As the sequences of whole cancer genomes become available, it is becoming clear that RAS proteins can be activated by missense mutations at amino acids other than the canonical sites (12, 13, and 61), or even via amplification or translocation of the wild-type29. Certain missense mutations at a given amino acid are more common than others and the frequency of any given mutation in a population depends on which cancer is under investigation. In some cases, the increased frequency of a specific allele can be explained by invoking a mutational mechanism – the G12C allele results from a classical smoking-induced mutation (G:C to T:A transversion) and is therefore enriched in lung cancer. This particular mutation has gained notoriety recently as two different labs have identified covalent inhibitors that attack the free cysteine and inhibit the activated oncoprotein30,31. This is the first example of an allele-specific therapy for RAS.
In other cases, the basis for allele enrichment is not as clear. For example, codon 146 mutations (typically A146T or A146V) are relatively common in CRC (they also occur at low frequency in AML), but have not been detected in cancers of the lung and pancreas, even though canonical activating mutations are common in those contexts32. A146T is a weakly activating mutation that promotes nucleotide exchange in the absence of guanine nucleotide exchange factor33. Since codon 146 mutations activate RAS through a molecular mechanism distinct from canonical mutations, there is a good possibility for allele-specific therapeutic opportunities. For example, Dafna Bar-Sagi and Paramjit Arora have generated peptides that bind to RAS and inhibit its interaction with SOS, therefore inhibiting SOS-induced nucleotide exchange34. Perhaps it would be possible to make a similar peptide that prevents SOS-independent nucleotide exchange in the codon 146 mutants, thereby locking them into a GDP-bound state. Alternatively, the unique mechanism of activation of codon 146 mutants could alter the way KRAS engages effector pathways, leading to differential sensitivity of inhibitors of downstream kinases. Indeed, experimental data indicate that CRC cell lines expressing KRASA146T are sensitive to MEK inhibition as a single agent, while cell lines expressing canonical KRAS activating mutations are not11. The molecular mechanism underlying this differential response is not known, and whether this observation will extend to mouse models and/or human patients remains to be seen.
Context
Everything I have discussed thus far is will almost certainly be highly dependent upon context. What does “context” mean? I would define context as everything except the RAS mutation itself. By this I mean the genetic, cellular, and physiologic context in which the RAS mutation arises. This is a somewhat esoteric definition, but context is not entirely a black box. Human cancer genetics can, again, offer some insight. For example, codon 146 mutations in KRAS are largely specific to CRC. What is different between CRC and cancers of the lung and pancreas, the other types of adenocarcinoma that common express mutant KRAS? To start, more than 90% of CRCs have activated Wnt signaling due to loss of APC or activation of beta-catenin. Back in our initial characterization of KRASG12D in the colonic epithelium, we noted that the hyperproliferative colon (which has normal Wnt signaling) was sensitive to MEK inhibition, while APC-mutant tumors (which have activated Wnt) expressing KRASG12D were not14. Similarly, NRAS mutations are more common in AML than in CRC, and mouse modeling revealed that NRASG12D affects basal homeostasis in myeloid cells, but not in the colon14,16. What makes the myeloid compartment special with respect to NRAS signaling is not clear.
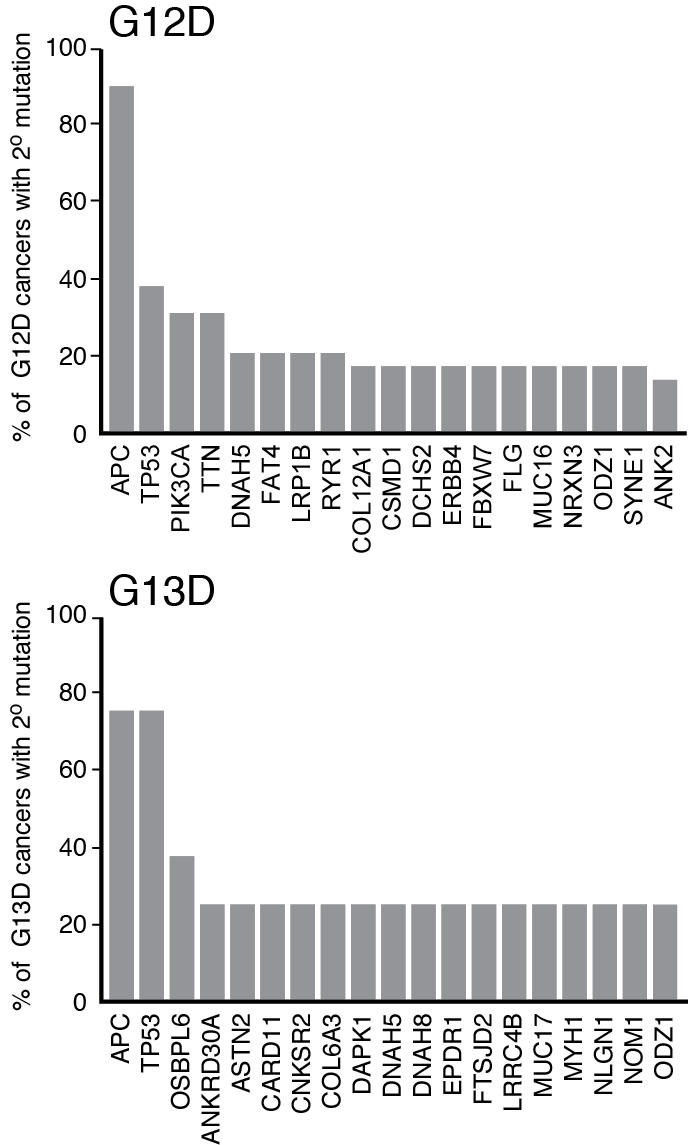
Figure 3. Secondary mutation profiles of CRCs expressing mutant KRAS. APC and TP53 are the most commonly mutated genes in cancers expressing oncogenic KRAS (regardless of allele), but beyond these two genes the secondary mutation profiles of cancers expressing KRASG12D and KRASG13D are largely different. Of note, PIK3CA and TTN mutations, which occur in more than 30% of CRCs expressing KRASG12D are absent in cancers expressing KRASG13D. Mutation data were obtained from The Cancer Genome Atlas.
It is important to note that allele effects and contextual effects are sometimes difficult to separate in human cancers because the secondary mutation profiles can be distinct. (The question of secondary mutations is addressed in the July 7, 2015 essay from Bob Stephens and Jim Hartley.) One example of this conundrum is the potential response of CRCs expressing KRASG13D to Cetuximab (mentioned above). Based on the published data from the TCGA, the other genes mutated in CRCs expressing KRASG13D are largely different from those expressing KRASG12D (Fig. 3). Most notably, PIK3CA mutations co-occur with KRASG12D in 30% of cases, but not at a detectable frequency in cancers expressing KRASG13D. PIK3CA mutations are associated with resistance to Cetuximab, so maybe the response to Cetuximab of CRCs expressing KRASG13D has nothing to do with the KRAS genotype, but rather stems from the lack of PIK3CA mutations in this population of cancers35. With the strongly likelihood that secondary mutations affect flux through RAS effector pathways and/or the response to inhibition of those pathways, precision medicine for KRAS will require detailed knowledge of the co-mutation profile, not only on a cancer-by-cancer basis, but also on an allele-by-allele basis. In terms of understanding how context affects the oncogenic properties of different RAS isoforms and different alleles, mouse models will undoubtedly be the most effective way to study this, as specific alleles can be generated and studied on a standardized genetic background.
Conclusion
As we learn more about the genetic events that contribute to cancer initiation and progression, it is becoming increasingly clear that knowing which individual genes are mutated in an individual’s cancer might not be enough to design effective personalized therapies (think Vemurafenib in BRAF mutant CRC). An alternative to the “one gene : one drug” idea is an approach in which we design distinct therapeutic strategies for each and every mutation within an individual gene, taking into account the context in which that mutant functions. RAS may be the poster child for this “be more precise” movement in the PMI. In the future, there may be dozens of distinct therapeutic approaches for “RAS mutant” cancers, taking into account (1) isoform, (2) mutation, (3) genetic context (i.e. secondary mutations), and (4) organ site.
About the Author
Kevin Haigis received his Ph.D. from the University of Wisconsin - Madison and did his post-doctoral work with Tyler Jacks at the Massachusetts Institute of Technology. Following his postdoctoral work, he began his independent career at the Massachusetts General Hospital. He is current the Director of Cancer Genetics at Beth Israel Deaconess Medical Center and an Associate Professor of Medicine at Harvard Medical School.