Faint Light at the End of the Tunnel
, by Gideon Bollag

Gideon Bollag, PhD
Drug development is fraught with obstacles, and developing drugs to block the RAS pathway becomes particularly challenging as highlighted in many past entries to this dialogue starting with Frank McCormick. Taking the perspective of a drug developer, I thought it appropriate to point out some less obvious challenges and then focus considerations on targeting the direct RAS effector RAF.
Perhaps the first risk involves target validation as very nicely illustrated in David Heimbrook’s entry. While clearly a compound that effectively blocks the RAS pathway will transform cancer treatment, it is important to recognize the more mundane details of drug discovery and development. An early obstacle of major importance is therapeutic index; RAS is a critical pathway for most normal cells so mutant selectivity is likely to be particularly important. Downstream effectors include much more druggable targets than mutant RAS itself such as RAF, MEK and ERK kinases, but introduce new challenges due to the complicated biology of the RAS pathway.
Once a target is chosen, the next challenge involves hit discovery and lead optimization. With recent technological improvements, many of the traditional hurdles in medicinal chemistry can now be overcome, though persistence is still a required virtue. For brevity, let’s assume a compound that meets profile criteria is selected for clinical development. Note that this includes appropriate translation from test tube to petri dish to animal model. A new series of potential pitfalls, less familiar to the academic scientist, now come to bear. Can the compound achieve appropriate bioavailability in preclinical species for efficacy and safety evaluation? Does the activity of a compound in animals even translate to similar or the same activity in humans? Can the compound be formulated to achieve bioavailability in humans? How is the compound metabolized? What are the characteristics of the major metabolites generated? And many more potential pitfalls can be exemplified.
Pending appropriate formulation and metabolic studies, compounds need to pass rigorous toxicology hurdles. In order to determine if the compound might be safe enough to test in clinical trials, careful preclinical pharmacology studies should identify drug exposures that predict efficacy. Typically, for oral compounds intended for daily dosing, four-week toxicology studies in two animal species (e.g. rats and dogs) are conducted. Compounds occasionally fail at this stage, when the therapeutic index is small, namely the exposure at which preclinical efficacy is observed exceeds that which is deemed tolerable in animal studies.
A few words on toxicity: it would not be surprising if compounds that block a central signaling pathway in almost all cells, i.e. the RAS pathway, would cause toxicity. However, this in itself should not be a deterrent, as multiple examples of inhibitors that block central pathways have made it through to the marketplace. Inhibitors of the RAF/MEK/ERK pathway are good examples. RAF inhibitors were the first therapeutics in this pathway to show clinical benefit1, 2. One might expect that isozyme selectivity, i.e. compounds specific for BRAF over CRAF or ARAF would be necessary for tolerability. But the first few compounds like vemurafenib and dabrafenib are not particularly isoform selective.
While this entry will not specifically address feedback effects of RAF/MEK/ERK inhibition, it is important to highlight that a variety of mechanisms have been shown to affect cellular responses to RAF and MEK inhibitors. Details of these mechanisms have been described and reviewed recently3, 4.
Much has been published about the paradoxical pathway activation that is caused by these first generation RAF inhibitors5-8. In cells whose RAS pathway is activated through RAS mutation or through upstream stimulation, these first generation RAF inhibitors lead to activation of MEK and ERK. Clearly, this is a surprising and unexpected feature of these compounds, and toxicities have been linked to this property, including cutaneous squamous cell carcinomas and other RAS driven tumors9, 10. But it should also be noted that the inability to shut down the RAS signaling pathway is likely key to the tolerability of these compounds. For example, MEK inhibitors have also demonstrated efficacy in BRAF mutant melanoma, yet the therapeutic effects, e.g. response rates, are lower than seen with the RAF inhibitors11. MEK inhibitors do not suffer from paradoxical pathway activation, yet the therapeutic index appears to be lower. Likely, this is because MEK inhibitors block RAS pathway signaling in normal cells more effectively than the first-generation RAF inhibitors. Different flavors of MEK inhibitors, in part differentiated by variable sensitivity to feedback, would have variable efficacy to block RAS signaling12, 13. A recent review provides more information about MEK inhibitors and their possible role in RAS-mediated tumors14.
Resistance to BRAF inhibitors most commonly involves reactivation of the RAF/MEK/ERK pathway, including through RAS mutations15-17. Consequently, dual BRAF/MEK inhibitor treatment has been tested and this approach has led to regulatory approval of two combinations so far18.
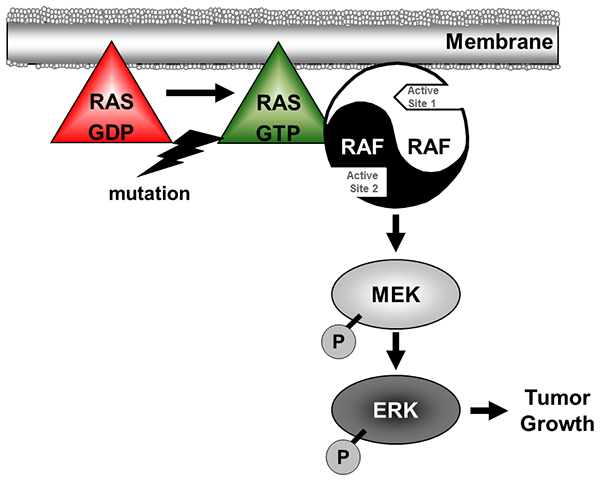
Figure 1. The role of RAF in the RAS pathway. Oncogenic mutation of RAS causes switch to the GTP-bound state which in turn recruits RAF dimers (or heterodimers) to the membrane. Dimeric RAF (further activated through phosphorylation events not shown here) then catalyzes activating phosphorylation of MEK which in turn catalyzes activating phosphorylation of ERK and leads to pro-tumor phenotypes. The asymmetric RAF dimer is illustrated in different colors to highlight the different conformations in the active sites.
Newer RAF inhibitors may effectively block the RAS pathway as well as (or better than) MEK inhibitors; some promising compounds have recently been identified19-21. These compounds derive from detailed mechanistic studies of the RAF inhibitor paradox. While a bit oversimplified, the following is a brief description of the mechanism: RAS induces RAF to dimerize (or hetero-dimerize) in a way that the two protomers of the dimer are asymmetric (adopt different conformations) 21-23. This mechanism is illustrated in Figure 1. First generation RAF inhibitors, including the older compound sorafenib24, have different affinities for the two asymmetric active sites, such that binding to one site allosterically activates the neighboring active site. One example of the kinase asymmetry involves the difference between type I and type II inhibitors. This distinction is partly defined by the activation state of the kinase: Type I inhibitors bind to an active state conducive to ATP-binding while type II inhibitors bind to a conformation that disfavors ATP binding25. Thus, broader RAF inhibition could be achieved by identifying a compound that binds with equal affinity to both asymmetric sites. A structural example illustrating this approach is shown in Figure 2.
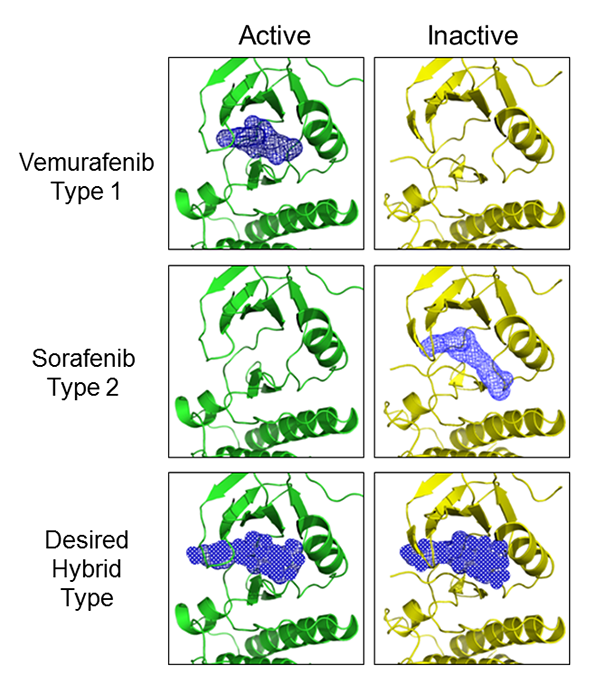
Figure 2. RAF binding modes. As visualized by x-ray crystallography, the RAF dimer often presents with one protomer in an active (green) and the other in an inactive (yellow) conformation. Co-crystallization of Type 1 inhibitors such as vemurafenib results in selective binding to the active conformation, while Type 2 inhibitors such as sorafenib selectively bind to the inactive conformation. A desired property of a drug that has improved potential to block RAS would adopt a hybrid binding mode.
Several different mechanisms can achieve this broader activity: selective inhibitors with equal affinity for the asymmetric sites21, co-targeting complementary kinases19, or a combination of these approaches20.
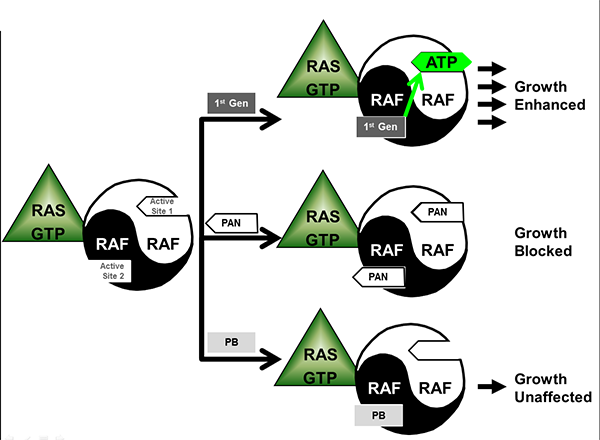
Figure 3. Variable effects of mechanistically-differentiated RAF inhibitors. Three different flavors of RAF inhibitor are schematically shown in this cartoon. First generation (1st Gen) inhibitors such as sorafenib, vemurafenib and dabrafenib bind to the active site of one protomer causing an allosteric activation on the adjacent protomer. Compounds that bind with similar affinities to the two asymmetric active sites (PAN) would block most signaling downstream of RAS. Compounds that bind selectively to one site but don’t cause allosteric activation of the adjacent protomer (‘paradox breaker’ or PB) do not affect signaling in a cell driven by mutant RAS.
At Plexxikon, we have adopted a different approach that does not block RAS, but rather that avoids the paradoxical activation in RAS-mutant cells. These compounds, dubbed ‘paradox breakers,’ bind to only one of the asymmetric sites, but do not allosterically activate the alternate site26. A clinical study is currently ongoing to test the hypothesis that such a paradox breaker, PLX8394, can be therapeutically active in BRAF-mutant cancers with less risk of the paradox-linked side-effects. A comparison of the various approaches for inhibiting the RAF dimer is shown in Figure 3.
Getting back to drug development, though, it is useful to understand additional pitfalls. One of the most common challenges involves achieving appropriate drug levels via an orally administered compound. Once ingested, the compound must be absorbed in the digestive tract without causing too much gastrointestinal distress; nausea, emesis and diarrhea are typical signs of this distress. Furthermore, since the typical protein kinase active site is largely hydrophobic, kinase inhibitors are often poorly soluble in aqueous media; poor solubility can lead to poor absorption. One illustration of this challenge is derived from the drug development of vemurafenib, one of the first-generation RAF inhibitors discovered at Plexxikon and developed in collaboration with Roche. An initial crystalline formulation achieved modest drug levels in the plasma of cancer patients, likely due to its poor solubility. Re-formulation by Roche yielded an amorphous material that enabled substantially (5-fold) better absorption that was critical for development of the drug27.
As described above, one promising approach to eventually block RAS signaling involves inhibiting RAS effectors such as RAF. Much has been learned over the last decade about the inhibition of RAF, yet we still have much to learn. The light is getting stronger, but the tunnel is long.
About the Author
Gideon Bollag earned his Ph.D. in biochemistry from Berkeley and his BS in chemistry from Penn State. He started his oncology career as a postdoc with Frank McCormick at Cetus Corporation. He then joined as one of the initial scientists when Frank founded Onyx Pharmaceuticals, where he eventually became senior director of small molecule therapeutics. He was involved in the Onyx collaborations to discover Nexavar and Ibrance, both now approved cancer treatments. Since 2002, Gideon has been with Plexxikon where he has led the discovery and development of the precision medicine Zelboraf. He is currently CEO of Plexxikon.