
Colaboramos con pacientes, investigadores y defensores de pacientes para mejorar la vida de jóvenes con cánceres raros
MyPART – Red sobre los Tumores Raros en Niños, Adolescentes y Jóvenes
-
 La Red MyPART
La Red MyPARTMyPART está formada por un grupo de científicos, pacientes, familiares, defensores de los pacientes y proveedores de atención de la salud que se centran en el estudio de tumores raros.
-
 Participe en nuestro ensayo
Participe en nuestro ensayoUsted puede ayudar a acelerar el desarrollo de nuevos tratamientos al proporcionar a los investigadores las herramientas que necesitan.
-
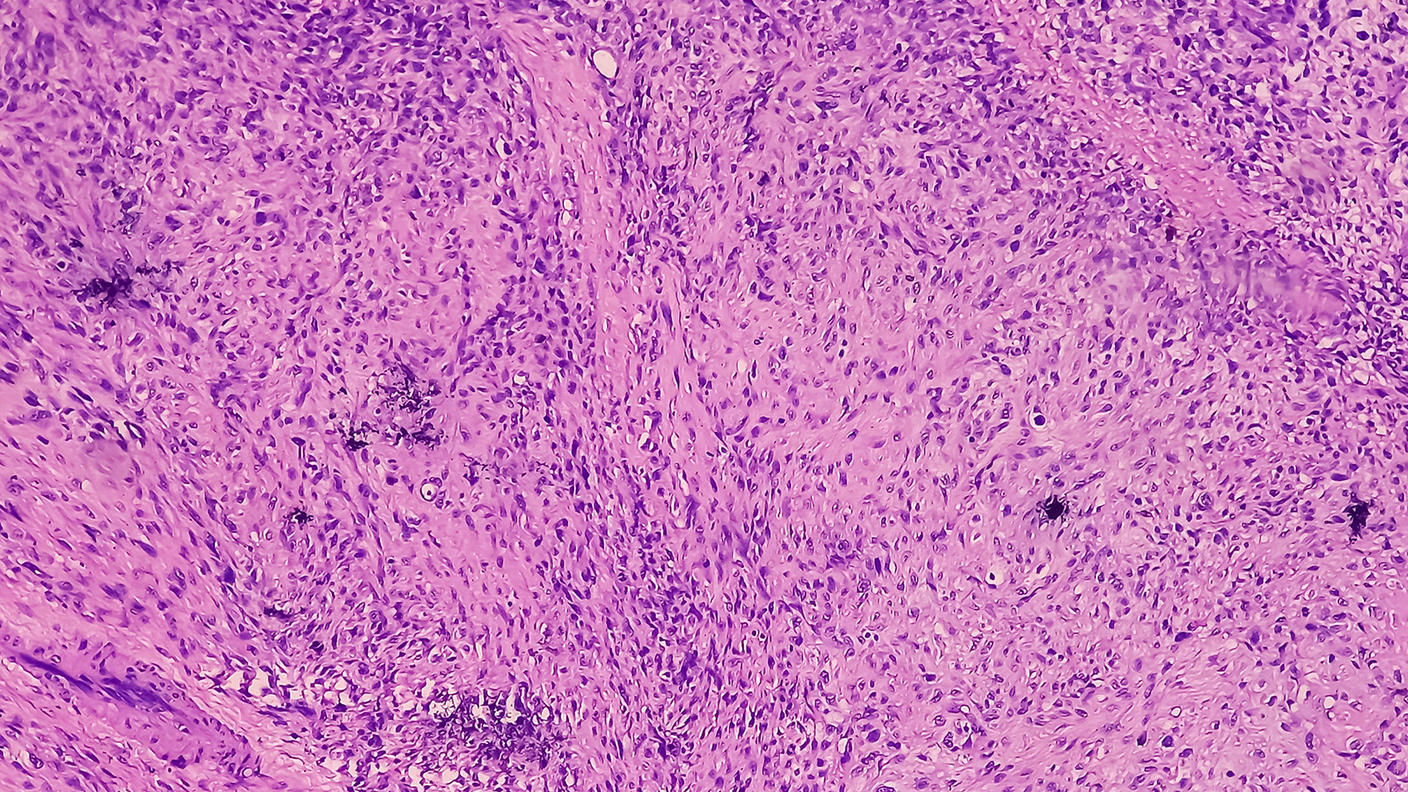 Por qué estudiamos los cánceres raros
Por qué estudiamos los cánceres rarosLos cánceres raros representan la cuarta parte de todos los cánceres. Descubra por qué los estudiamos.

El nirogacestat ofrece esperanza a las personas con tumores desmoides
El tratamiento con nirogacestat achicó los tumores, mejoró la supervivencia sin progresión y alivió el dolor.
Más