Durante las últimas 5 décadas, se han logrado avances notables en la formulación de tratamientos curativos para las neoplasias malignas infantiles. Se espera que más del 80 % de los niños con cáncer sobrevivan hasta la edad adulta si tienen acceso a tratamientos contemporáneos.[1] En ocasiones, los tratamientos que hacen posible esta supervivencia también producen desenlaces adversos a largo plazo para la salud, conocidos como efectos tardíos, que se manifiestan meses o años después de terminar el tratamiento del cáncer.
Se han utilizado muchos abordajes para estudiar la morbilidad a muy largo plazo relacionada con el cáncer infantil y su contribución a la mortalidad temprana. En estas iniciativas se ha usado una gama de recursos que incluye datos de las siguientes fuentes:
Se necesitan datos de gran calidad para establecer los perfiles de presentación y riesgo de toxicidad tardía del tratamiento del cáncer; por lo general, esos datos provienen de estudios en los que se notifican desenlaces en sobrevivientes sometidos a evaluaciones médicas que proporcionan estados clínicos en detalle, exposiciones al tratamiento y efectos tardíos específicos. Cualquiera que sea la metodología de un estudio, es importante tener en cuenta el sesgo de selección y de participación en los estudios de cohortes en el contexto de los resultados.
Prevalencia de los efectos tardíos en los sobrevivientes de cáncer infantil
Por lo general, se presentan efectos tardíos en adultos que sobrevivieron al cáncer infantil; la prevalencia de los efectos tardíos aumenta con el transcurso del tiempo desde el diagnóstico del cáncer. En estudios multinstitucionales y poblacionales, se observó un riesgo excesivo de morbilidad hospitalaria en sobrevivientes de cáncer diagnosticado durante la niñez o juventud, en comparación con los controles ajustados por edad y sexo, con alguna evidencia de que este riesgo es desproporcionadamente alto entre los sobrevivientes de poblaciones raciales y étnicas minoritarias.[3,9-13]
Entre los adultos tratados por cáncer durante la niñez, los efectos tardíos contribuyen a una carga de morbilidad alta. En las investigaciones se observó lo siguiente:[4,7,14-18]
Del 60 % a más del 90 % de los sobrevivientes presentan una o más afecciones crónicas.
Del 20 % al 80 % de los sobrevivientes experimentan complicaciones graves o potencialmente mortales en la edad adulta.
La acumulación de morbilidad se acelera en los adultos jóvenes sobrevivientes de cáncer infantil, en comparación con la de sus hermanos y la de la población general. La acumulación de enfermedades crónicas predice el riesgo de muerte temprana.[19]
La finalidad del estudio de cohorte St. Jude Life (SJLIFE) fue describir la carga acumulada del tratamiento del cáncer mediante un parámetro de carga acumulada, que incorpora múltiples afecciones y episodios de recidiva en un solo parámetro que tiene en cuenta riesgos contrapuestos. A los 50 años de edad, los sobrevivientes de esta cohorte presentaron un promedio de 17,1 afecciones crónicas, 4,7 de las cuales fueron graves o incapacitantes, potencialmente mortales o mortales.[16] Este hallazgo contrasta con la carga acumulada en controles emparejados de la comunidad, que presentaron 9,2 afecciones crónicas, 2,3 de las cuales fueron graves o incapacitantes, potencialmente mortales o mortales (consultar la Figura 1).[16]
AmpliarFigura 1. La figura muestra la distribución de la carga acumulada por edad entre los sobrevivientes de subtipos específicos de cáncer infantil y los controles comunitarios que participaron en el estudio de cohorte SJLIFE. La carga acumulada a los 30 años y la tasa de aumento de la carga acumulada varía entre los subtipos de cáncer y de sistemas orgánicos. Reproducción de The Lancet, volumen 390, número 10112, Bhakta N, Liu Q, Ness KK, Baassiri M, Eissa H, Yeo F, Chemaitilly W, Ehrhardt MJ, Bass J, Bishop MW, Shelton K, Lu L, Huang S, Li Z, Caron E, Lanctot J, Howell C, Folse T, Joshi V, Green DM, Mulrooney DA, Armstrong GT, Krull KR, Brinkman TM, Khan RB, Srivastava DK, Hudson MM, Yasui Y, Robison LL, The cumulative burden of surviving childhood cancer: an initial report from the St Jude Lifetime Cohort Study (SJLIFE), páginas 2569–2582, Derechos de autor (2017), autorizada por Elsevier. Cumulative burden of individual: carga acumulada individual; Neoplasms: neoplasias; Cardiovascular: cardiovascular; Renal: renal; Haematological: hematológico; Ocular: ocular; Gastrointestinal: gastrointestinal; Pulmonary: pulmonar; Endocrine: endocrino; Musculoskeletal: osteomuscular; Neurological: neurológico; Reproductive: reproductor; Infections: infecciones; Auditory: auditivo; Controls: controles; ALL: leucemia linfoblástica aguda; AML: leucemia mieloide aguda; Hodgkin's: linfoma de Hodgkin; NHL: linfoma no Hodgkin; CNS: sistema nervioso central; Bone tumours: tumores óseos; STS: sarcomas de tejidos blandos; Wilms' tumours: tumor de Wilms; Neuroblastoma: neuroblastoma; Retinoblastoma: retinoblastoma; Germ cell: células germinativas; Primary diagnosis by age strata: diagnóstico primario por estrato de edad.
Los investigadores del estudio de cohorte SJLIFE compararon la carga acumulada de afecciones crónicas en 4612 sobrevivientes adolescentes y adultos jóvenes a la edad de 18 años (período de transición de la atención pediátrica a la atención como adultos dentro del sistema de salud) y a la de 26 años (período de transición del plan de seguro familiar a un plan de seguro individual) con la de 625 controles.[20]
A la edad de 18 años, los sobrevivientes padecieron en promedio 22,3 afecciones incapacitantes por cada 100 personas versus 3,5 en los controles, y también padecieron 128,7 afecciones de menor gravedad (en riesgo de progresar a una afección con mayor grado de incapacidad) versus 12,4 en los controles.
A la edad de 26 años, los sobrevivientes padecieron en promedio 40,3 afecciones incapacitantes por cada 100 personas versus 5,7 en los controles, y también padecieron 240,5 afecciones de menor gravedad versus 51,3 en los controles.
La carga acumulada de afecciones específicas causantes de discapacidad a la edad de 18 y 26 años fue más sobresaliente en los sobrevivientes de tumores óseos (osteomusculares: 99,9 y 121,70, respectivamente), sarcomas de tejido blando (osteomusculares: 49,5 y 54,1, respectivamente), y tumores del sistema nervioso central (SNC) (neurológicos: 24,7 y 36,8, respectivamente).
La carga acumulada de afecciones de menor gravedad (susceptibles de intervención) a los 18 y 26 años fue más notable para las afecciones neurológicas en la mayoría de subgrupos de cáncer; la carga acumulada más alta fue para los sobrevivientes de tumores del SNC (95,2 y 162,3, respectivamente).
Estos hallazgos resaltan la importancia de optimizar el acceso a la atención médica y a los seguros médicos para los sobrevivientes a medida que envejecen y ya no les es posible recibir atención en los sistemas de atención pediátrica.
La variabilidad de la prevalencia se relaciona con diferencias en las siguientes características:
Edad y período de seguimiento de las cohortes estudiadas.
Métodos y congruencia de la evaluación (por ejemplo, autonotificación vs. evaluaciones médicas según el riesgo).
Intensidad y época del tratamiento.
Los investigadores del Childhood Cancer Survivor Study (CCSS) demostraron que el riesgo elevado de morbilidad y mortalidad en los sobrevivientes de la cohorte, a medida que envejecen, aumenta más allá de la 4.ᵃ década de vida. A los 50 años, la incidencia acumulada autonotificada de una afección grave, incapacitante, potencialmente mortal o mortal fue del 53,6 % en los sobrevivientes, en comparación con el 19,8 % en los hermanos (de ambos sexos) del grupo de control. Entre los sobrevivientes que llegaron a la edad de 35 años sin presentar ninguna afección previa grave, incapacitante, potencialmente mortal o mortal, el 25,9 % presentó una nueva afección de grado 3 a grado 5 dentro de los 10 años siguientes, en comparación con el 6,0 % de los hermanos sanos.[4]
La presencia de afecciones crónicas graves, incapacitantes y potencialmente mortales perjudica el estado de salud de los sobrevivientes que envejecen, sobre todo en lo relativo al deterioro funcional y las limitaciones de las actividades. Como era previsible, se ha notificado que las afecciones crónicas contribuyen a una prevalencia más alta de síntomas de sufrimiento emocional en los adultos sobrevivientes que en los controles poblacionales.[21] Las mujeres sobrevivientes exhiben un deterioro del estado de salud en función de la edad más pronunciado que los varones sobrevivientes.[22] La prevalencia aún más alta de efectos tardíos entre las cohortes analizadas usando criterios clínicos, se relaciona con las afecciones subclínicas y no diagnosticadas que se identifican mediante exámenes de detección y estrategias de vigilancia.[7]
Los investigadores del CCSS también evaluaron el efecto de la raza y la etnia en los desenlaces tardíos. En el estudio se comparó la mortalidad tardía, las neoplasias subsiguientes y las afecciones crónicas de los participantes hispanos (n = 750) y participantes negros que no son hispanos (n = 694) con los participantes blancos que no son hispanos (n = 12 397).[23] Se observaron los siguientes resultados:
El tratamiento del cáncer no explicó las desigualdades en la mortalidad, las afecciones crónicas ni las neoplasias subsiguientes observadas entre los grupos.
Las diferencias en el nivel socioeconómico y los factores de riesgo cardiovascular afectaron el riesgo. La mortalidad por todas las causas fue más alta en los participantes negros que no son hispanos que en los otros grupos, pero esta diferencia desapareció después del ajuste según el nivel socioeconómico.
El riesgo de presentar diabetes fue elevado entre los grupos de minorías raciales o étnicas, incluso después del ajuste por nivel socioeconómico y obesidad.
Fue más probable que los pacientes negros que no son hispanos notificaran afecciones cardíacas, pero este riesgo disminuyó después del ajuste por factores de riesgo cardiovascular.
Los participantes negros que no son hispanos no notificaron cáncer de piel no melanoma, un hallazgo reproducido en otros estudios;[24] los participantes hispanos tuvieron un riesgo más bajo de presentar cáncer de piel no melanoma que los participantes blancos no hispanos.
El reconocimiento de los efectos tardíos, junto con los avances en la biología del cáncer, las ciencias radiológicas y los cuidados de apoyo, ha dado lugar a un cambio en la prevalencia y variedad de los efectos del tratamiento. En un esfuerzo por reducir y prevenir los efectos tardíos, el tratamiento contemporáneo de la mayoría de las neoplasias malignas infantiles ha evolucionado hacia un enfoque adaptado al riesgo, que se asigna de acuerdo con una variedad de factores clínicos, biológicos y, a veces, genéticos.
En el CCSS se notificó que, con la disminución de la dosis acumulada y la frecuencia de la radiación terapéutica de 1970 a 1999, los sobrevivientes experimentaron una disminución significativa del riesgo de neoplasias subsiguientes.[25]
A excepción de los sobrevivientes que necesitan tratamiento multimodal intensivo para neoplasias malignas resistentes al tratamiento o en recaída, los efectos potencialmente mortales del tratamiento son relativamente poco comunes luego de los tratamientos contemporáneos durante el seguimiento temprano (hasta 10 años después del diagnóstico).
Sin embargo, con frecuencia, los sobrevivientes aún presentan una morbilidad que les altera la vida debido a los efectos del tratamiento del cáncer en el funcionamiento endocrino, reproductivo, osteomuscular y neurológico.
En una investigación del CCSS se examinaron los patrones temporales de incidencia acumulada de afecciones crónicas graves y mortales en los sobrevivientes tratados entre 1970 y 1999.[26]
La incidencia acumulada a 20 años de por lo menos una afección crónica de grado 3 a 5 disminuyó de manera significativa, del 33,2 % para los sobrevivientes que recibieron el diagnóstico entre 1970 y 1979, al 29,3 % para los que recibieron el diagnóstico entre 1980 y 1989, y hasta el 27,5 % para los que recibieron el diagnóstico entre 1990 y 1999, en comparación con una incidencia del 4,6 % en la cohorte de hermanos.
La disminución general en la incidencia de afecciones crónicas durante las 3 décadas de tratamiento se explica, en parte, por la disminución sustancial de las endocrinopatías, las neoplasias malignas subsiguientes (NMS), las afecciones osteomusculares y las afecciones gastrointestinales, mientras que la incidencia acumulada de hipoacusia aumentó durante este período.
La disminución en la morbilidad no fue uniforme en los diferentes grupos de diagnóstico o tipos de afección debido a diferencias en los patrones de tratamiento y supervivencia a lo largo del tiempo. Para obtener más información en inglés, consultar la Figura 2.
A pesar de la disminución de las afecciones crónicas a lo largo del tiempo, el estado de salud autonotificado no ha mejorado en las épocas de tratamiento más recientes. Es posible que este hallazgo se deba a la supervivencia de los niños con enfermedad de riesgo más alto que habrían fallecido de cáncer en épocas anteriores, o a un conocimiento más profundo de los efectos tardíos y su vigilancia entre los sobrevivientes tratados de manera más reciente.[27]
AmpliarFigura 2. Incidencia acumulada de enfermedades crónicas de grado 3 a 5 en sobrevivientes de cáncer infantil a 5 años por década de diagnóstico e incidencia en hermanos. A) Incidencia acumulada de la primera afección de grado 3 a 5. B) Incidencia acumulada de 2 o más afecciones de grado 3 a 5. El área sombreada representa el intervalo de confianza (IC) de 95 %. Debajo del eje x se indica el número de participantes en riesgo (número censurado) en cada intervalo de 5 años posterior al diagnóstico. El número censurado no incluye a los que presentaron un episodio competitivo de riesgo (muerte por una causa diferente a una afección crónica de grado 5). Reproducción de The Lancet Oncology, volumen 19, número 12, Todd M Gibson, Sogol Mostoufi-Moab, Kayla L Stratton, Wendy M Leisenring, Dana Barnea, Eric J Chow, Sarah S Donaldson, Rebecca M Howell, Melissa M Hudson, Anita Mahajan, Paul C Nathan, Kirsten K Ness, Charles A Sklar, Emily S Tonorezos, Christopher B Weldon, Elizabeth M Wells, Yutaka Yasui, Gregory T Armstrong, Leslie L Robinson, Kevin C Oeffinger, Temporal patterns in the risk of chronic health conditions in survivors of childhood cancer diagnosed 1970–99: a report from the Childhood Cancer Survivor Study cohort. Páginas 1590-1601, Derechos de autor (2018), autorizada por Elsevier. Cumulative Incidence (%): incidencia acumulada (%); Siblings: hermanos; Time since diagnosis (years): tiempo desde el diagnóstico (años); Number at risk (number censored): número en riesgo (número censurado).
Mortalidad
Tal como se observa en los siguientes estudios, los efectos tardíos también contribuyen a un exceso de riesgo de muerte prematura entre los sobrevivientes de cáncer infantil a largo plazo:
En varios estudios de cohortes grandes de sobrevivientes, se notificó mortalidad temprana en las personas tratadas por cáncer infantil en comparación con los controles de la población general emparejados por edad y sexo. El cáncer primario en recaída o resistente al tratamiento sigue siendo la causa más frecuente de muerte, seguido de un exceso de mortalidad por causa específica debido a cánceres primarios subsiguientes, así como toxicidad cardíaca y pulmonar.[28-33]
En un estudio del CCSS, se evaluaron las causas específicas de mortalidad tardía y exceso de muertes por causas relacionadas con la salud, en comparación con la población general de los Estados Unidos para identificar objetivos que permitan reducir el riesgo futuro.[33]
Al cabo de una mediana de seguimiento de 29 años desde el diagnóstico, la tasa acumulada de mortalidad a 40 años por todas las causas fue del 23,3 %, y el 51,2 % de las muertes se atribuyeron a causas relacionadas con la salud.
A partir de 40 años o más desde el diagnóstico, se produjo un exceso de 131 muertes de sobrevivientes relacionadas con la salud por 10 000 años-persona (intervalo de confianza [IC] 95 %, 111–163). El exceso de muertes se relacionó con mayor frecuencia con cáncer (exceso de riesgo absoluto por 10 000 años-persona, 54; IC 95 %, 41–68), cardiopatía (27; 18–38) y enfermedad cerebrovascular (10; 5–17).
El estilo de vida saludable (evaluado por el consumo de tabaco y de bebidas alcohólicas, la actividad física, el índice de masa corporal y la ausencia de hipertensión y diabetes) se relacionó con una reducción del 20 % al 30 % en la mortalidad relacionada con la salud, con independencia de otros factores.
En un análisis de los datos del CCSS y del Surveillance, Epidemiology, and End Results (SEER) Program se evaluó la supervivencia condicional. En el estudio se demostró una tasa de supervivencia subsiguiente a 5 años del 92 % o más en la mayoría de los diagnósticos a los 5, 10, 15 y 20 años. Entre las personas que sobrevivieron por lo menos 5 años a partir del diagnóstico, la probabilidad de mortalidad por todas las causas en los siguientes 10 años fue del 8,8 % en el CCSS y del 10,6 % en el estudio SEER, y las neoplasias representaron la causa de muerte en alrededor del 75 % de los sobrevivientes.[34]
A pesar de las tasas altas de morbilidad prematura, la mortalidad general ha disminuido con el tiempo.[28,30,31,35,36]
Los investigadores del CCSS evaluaron la mortalidad tardía por cualquier causa y la mortalidad tardía relacionada con la salud (incluso con efectos tardíos del tratamiento del cáncer), las NMS, las afecciones crónicas y los desenlaces neurocognitivos entre 6148 sobrevivientes de leucemia linfoblástica aguda infantil (mediana de edad, 27,9 años; intervalo, 5,9–61,9 años) diagnosticados entre 1970 y 1999.[37]
En términos generales, la tasa de mortalidad tardía por cualquier causa a 20 años fue del 6,6 %.
En comparación con los participantes que se trataron en la década de 1970, los participantes que recibieron tratamiento con regímenes estratificados según el riesgo en la década de 1990 presentaron una mortalidad tardía relacionada con la salud más baja (razón de tasas: riesgo estándar en la década de 1990, 0,2; riesgo alto en la década de 1990, 0,3), lo que fue comparable con la población de los Estados Unidos (razón de mortalidad estandarizada [RME]: riesgo estándar en la década de 1990, 1,3; riesgo alto en la década de 1990, 1,7).
El riesgo de mortalidad tardía y afecciones crónicas graves ha disminuido con el tiempo entre los sobrevivientes de leucemia mieloide aguda (LMA). Los investigadores del CCSS evaluaron la morbilidad, mortalidad y estado de salud a largo plazo a 5 años de más de 800 de sobrevivientes de LMA infantil según el tratamiento y la época del tratamiento. Los sobrevivientes se compararon por grupo de tratamiento (trasplante de células madre hematopoyéticas [TCMH]); quimioterapia con radiación craneal [RTC]; quimioterapia sola) y década de diagnóstico.[38]
Entre 856 sobrevivientes, la incidencia acumulada de mortalidad tardía a 20 años fue más alta después del TCMH (13,9 %; quimioterapia con RTC, 7,6 %; quimioterapia sola, 5,1 %).
La incidencia acumulada de mortalidad disminuyó en los sobrevivientes de TCMH diagnosticados en la década de 1990 (8,5 %), en comparación con la década de 1970 (38,9 %), al igual que las tasas de mortalidad estandarizadas.
El estado de salud autonotificado fue de bueno a excelente para el 88,2 % de los sobrevivientes de LMA infantil, con independencia del tratamiento.
La mayoría de los sobrevivientes no presentaron ninguna afección crónica de grado 3 a 5 después de 20 años (TCMH, 45,8 %; quimioterapia con RTC, 23,7 %; quimioterapia sola, 27,0 %).
Se observó una reducción temporal de la incidencia acumulada de afecciones crónicas después del TCMH (1970, 76,1 %; 1990, 38,3 %; P = 0,02), reflejo de una reducción en el uso de irradiación corporal total.
Se utilizaron datos poblacionales de un registro estatal de cáncer para evaluar las diferencias en la supervivencia y los desenlaces a largo plazo por raza y etnia en 4222 niños con diagnóstico de cáncer entre 1987 y 2012.[12]
En comparación con los niños blancos que no son hispanos, la hospitalización fue un 70 % (cociente de riesgos instantáneos [CRI], 1,7) más común 5 o más años después del diagnóstico para los niños indígenas de los Estados Unidos y nativos de Alaska y un 50 % (CRI, 1,5) más común para los niños negros.
Entre los sobrevivientes a los 5 o más años del diagnóstico, se observaron aumentos relativos estadísticamente significativos de 2,3 a 3,6 veces en las hospitalizaciones por afecciones específicas para niños indígenas de los Estados Unidos y nativos de Alaska (CRI, 2,3 para afecciones relacionadas infecciones; CRI, 3,0 para afecciones hematológicas; CRI, 2,6 para afecciones digestivas). Los mayores aumentos se observaron en las afecciones relacionadas con la salud mental (CRI, 3,6), un patrón que también se observó en los niños negros (CRI, 2,5).
En un análisis del estudio de cohorte SJLIFE se exploraron las relaciones entre las afecciones crónicas modificables y la mortalidad tardía en el contexto de los determinantes sociales de la salud.[39]
Entre los 9440 sobrevivientes a 5 años de cáncer infantil que se incluyeron en el análisis (mediana de edad en el momento del último seguimiento, 27,5 años; mediana de seguimiento, 18,8 años), la mortalidad por todas las causas (RME, 7,6; IC 95 %, 7,2–8,1) y la mortalidad tardía relacionada con la salud (RME, 7,6; IC 95 %, 7,0–8,2) fue significativamente más alta de lo esperado para las tasas de mortalidad en los Estados Unidos.
Entre los 3407 participantes adultos que completaron una evaluación en el campus (mediana de edad en el momento de la evaluación, 35,4 años; mediana de seguimiento, 27,3 años), se relacionaron los aumentos significativos en las muertes tardías por todas las causas y las vinculadas con la salud con el número de afecciones crónicas modificables, el pertenecer a una sección del censo de los Estados Unidos asociado con un índice de privación de área alto y fragilidad.
Las asociaciones específicas para el exceso de mortalidad relacionada con la salud fueron las siguientes: 1 afección crónica modificable de grado 2 (razón de tasas [RR], 2,2; IC 95 %, 1,1–4,4), 2 afecciones crónicas modificables de grado 2 (RR, 2,5; IC 95 %, 1,2–5,2), 3 afecciones crónicas modificables de grado 2 (RR, 4,0; IC 95 %, 1,9–8,4), índice de privación de área en el percentil 51º a 80º (RR, 9,2; IC 95 %, 1,2–69,7), índice de privación de área en el percentil 81º a 100º (RR, 16,2; IC 95 %, 2,1–123,7) y fragilidad (RR, 2,3; IC 95 %, 1,2–4,1).
En el CCSS y en un estudio de cohorte del SJLIFE se investigó la contribución al riesgo de mortalidad tardía por NMS (5 años o más después del diagnóstico) de las variantes de predisposición al cáncer.[40]
De 12 469 participantes (6172 hombres y 6297 mujeres), entre los que se incluyeron 4402 de la cohorte del SJLIFE (mediana del tiempo de seguimiento desde la recolección de la primera muestra biológica, 7,4 años) y 8067 de la cohorte del CCSS (mediana del tiempo de seguimiento desde la recolección de la primera muestra biológica, 12,6 años), 641 (5,1 %) participantes presentaban variantes de predisposición al cáncer.
Las variantes de predisposición se relacionaron significativamente con un aumento de la gravedad de las NMS (criterios terminológicos comunes para el grado de efectos adversos ≥4 vs. grado <4: oportunidad relativa [OR], 2,15; IC 95 %, 1,18–4,19).
Se presentaron muertes relacionadas con las NMS en 263 participantes (2,1 %) y muertes por otra causa en 426 sobrevivientes (3,4 %).
Al cabo de 10 años desde de la recolección de la primera muestra biológica, la tasa de mortalidad acumulada relacionada con una NMS en los portadores de variantes de predisposición al cáncer fue del 3,7 % (IC 95 %, 1,2–8,5 %) en SJLIFE y del 6,9 % (4,1–10,7 %) en el CCSS. En comparación, la tasa de mortalidad acumulada relacionada con una NMS en aquellos que no presentaban variantes de predisposición al cáncer fue del 1,5 % (1,0–2,1 %) en el SJLIFE y del 2,1 % (1,7–2,5 %) en el CCSS.
La presencia de variantes de predisposición al cáncer se relacionó con un aumento del riesgo de muerte por NMS (SJLIFE: CRI, 3,40; IC 95 %, 1,37–8,43; CCSS: CRI, 3,58; IC 95 %, 2,27–5,63).
Sobrevivientes de cánceres diagnosticados durante la adolescencia y adultez temprana
Se dispone de poca información sobre la mortalidad tardía de los sobrevivientes de un cáncer diagnosticado cuando eran adolescentes o adultos jóvenes (AAJ).[41-44]
Con los datos del SEER, se estudió la supervivencia relativa condicional hasta 25 años después del diagnóstico en una cohorte de pacientes (N = 205 954) a los que se diagnosticó un primer cáncer maligno durante la adolescencia o adultez temprana (tiroides, melanoma, testículo, mama, linfoma, leucemia o tumores del SNC).[41]
Para todos los tipos de cáncer combinados, entre las personas que sobrevivieron hasta 5 años, la tasa relativa de supervivencia a 5 años subsiguiente fue superior al 95 % a los 7 años del diagnóstico.
La mayoría de los pacientes con cáncer diagnosticado durante la adolescencia o adultez temprana que sobrevivieron por lo menos 7 años después del diagnóstico presentaron pocas diferencias en la supervivencia, en comparación con la población general.
Para tipos de cáncer específicos, incluso tumores del SNC, cáncer de mama en mujeres, linfoma de Hodgkin y leucemia, existe evidencia de que el exceso de riesgo de mortalidad persistió o reapareció más de 10 años después del diagnóstico de cáncer.
La supervivencia relativa condicional fue más baja en pacientes AAJ, aunque los pacientes de 15 a 29 años exhibieron una tasa de supervivencia más alta que los pacientes de 30 a 39 años en el momento del diagnóstico de sus tumores del SNC.
En un análisis independiente de sobrevivientes a 5 años de cáncer diagnosticado durante la adolescencia o adultez temprana (edad 15–39 años en el momento del diagnóstico), donde también se utilizaron los datos del SEER (N = 282 969), se demostró lo siguiente:[42]
La tasa de mortalidad por cualquier causa a 10 años disminuyó desde el 8,3 % para los pacientes diagnosticados entre 1975 y 1984 hasta el 5,4 % para los pacientes diagnosticados entre 2005 y 2011.
La reducción de la mortalidad fue resultado, ante todo, de un menor número de muertes por el cáncer inicial.
Los investigadores del CCSS compararon las afecciones crónicas y la mortalidad por todas las causas y causas específicas entre 5804 sobrevivientes de cáncer de aparición temprana durante la adolescencia o adultez temprana (diagnóstico de cáncer, edad de 15–20 años; mediana de edad, 42 años) y 5804 sobrevivientes de cáncer infantil (diagnóstico de cáncer, edad <15 años; mediana de edad, 34 años) emparejados según el diagnóstico del cáncer primario.[43]
La RME fue de 5,9 (IC 95 %, 5,5–6,2) para los sobrevivientes de cáncer de inicio temprano durante la adolescencia o adultez temprana, y de 6,2 (IC 95 %, 5,8–6,6) para los sobrevivientes de cáncer infantil más jóvenes, en comparación con la población general.
Los sobrevivientes de cáncer de inicio temprano durante la adolescencia o adultez temprana tuvieron una RME más baja para la muerte por causas relacionadas con la salud que los sobrevivientes de cáncer infantil (RME, 4,8 [IC 95 %, 4,4–5,1] vs. 6,8 [IC 95 %, 6,2–7,4]), que se observó sobre todo más de 20 años después del diagnóstico de cáncer.
Los sobrevivientes de cáncer infantil y de cáncer de inicio temprano durante la adolescencia o adultez temprana tuvieron un riesgo mayor de presentar afecciones graves e incapacitantes, potencialmente mortales o mortales (grados 3 a 5) que hermanos y hermanas de la misma edad (CRI, 4,2 [IC 95 %, 3,7–4,8] para sobrevivientes de cáncer de inicio temprano durante la adolescencia o adultez temprana, y 5,6 [IC 95 %, 4,9–6,3] para sobrevivientes de cáncer infantil), si bien el riesgo fue menor para los sobrevivientes de cáncer de inicio temprano que para los sobrevivientes de cáncer infantil.
En un estudio de cohorte poblacional retrospectivo de Kaiser Permanente se examinó la mortalidad por causa específica en sobrevivientes a 2 años (n = 10 574) de cánceres diagnosticados durante la adolescencia o adultez temprana (pacientes de 13 a 39 años diagnosticados entre 1990 y 2012) y se comparó con la de personas sin cáncer.[45]
Los sobrevivientes de cáncer durante la adolescencia o adultez temprana tuvieron un riesgo de muerte 10,4 veces mayor, en comparación con la cohorte emparejada sin cáncer, y este riesgo siguió siendo elevado más de 20 años después del diagnóstico (razón de tasas de incidencia [RTI], 2,9).
A partir de los 15 años después del diagnóstico, la incidencia de la mortalidad relacionada con segundos cánceres superó la tasa de mortalidad relacionada con la recidiva.
El riesgo de muerte por suicidio se duplicó en los sobrevivientes de cáncer durante la adolescencia o adultez temprana, en comparación con la cohorte sin cáncer.
En un estudio de cohorte poblacional retrospectivo se investigaron las afecciones comórbidas a los 2 años del diagnóstico en 6778 sobrevivientes de cáncer diagnosticado durante la adolescencia o adultez temprana controlados en Kaiser Permanente.[46]
Alrededor del 17 % de los sobrevivientes presentaron más de una afección comórbida. Las afecciones comórbidas más comunes fueron dislipidemia (22 por 1000 años-persona), hipertensión (16 por 1000 años-persona), diabetes (10 por 1000 años-persona), trastornos tiroideos (9 por 1000 años-persona) y depresión o ansiedad graves (8 por 1000 años-persona).
Los RTI fueron más altos en los sobrevivientes que en los controles sin un antecedente de cáncer; las mayores diferencias se observaron en la necrosis avascular (RTI, 8,25), seguida por la osteoporosis (RTI, 5,75), el reemplazo articular (RTI, 3,89), el accidente cerebrovascular (RTI, 3,19), la insuficiencia ovárica prematura (CTI, 2,87), y la miocardiopatía o insuficiencia cardíaca (RTI, 2,64).
Para los sobrevivientes de cáncer en AAJ, la prevalencia de múltiples afecciones comórbidas fue de cerca del 40 % 10 años después de la fecha índice (punto de tiempo a los 2 años del diagnóstico), en comparación con el 20 % para aquellos sin cáncer (P < 0,001).
Vigilancia de los efectos tardíos
El reconocimiento tanto de la toxicidad aguda como de la específica tardía alentó las investigaciones para evaluar los factores fisiopatológicos y pronósticos de los efectos del tratamiento del cáncer. En consecuencia, los resultados de la investigación sobre los efectos tardíos han desempeñado una función importante en las siguientes áreas:
Cambio de los abordajes terapéuticos del cáncer infantil para reducir la mortalidad relacionada con el tratamiento en los sobrevivientes tratados en épocas más recientes.[47]
Formulación de recomendaciones para el asesoramiento sobre los riesgos y los exámenes médicos de detección para los sobrevivientes a largo plazo mediante la identificación de características clínicas y terapéuticas de quienes tienen un riesgo más alto de complicaciones del tratamiento.[48]
Los efectos tardíos comunes de los cánceres infantiles abarcan varios dominios amplios como los siguientes:
Crecimiento y desarrollo.
Funcionamiento orgánico.
Capacidad reproductiva y estado de salud de los descendientes.
Carcinogénesis secundaria.
Secuelas psicosociales relacionadas con el cáncer primario, su tratamiento o la falta de adaptación relacionada con la vivencia del cáncer.
Las secuelas tardías del tratamiento del cáncer infantil se pueden anticipar según las exposiciones terapéuticas, pero hay numerosos factores que influyen en la magnitud del riesgo y las manifestaciones en cada paciente. Se deben considerar múltiples factores en la evaluación del riesgo para un efecto tardío determinado (consultar la Figura 3).[49]
Factores relacionados con el cáncer
Órganos o tejidos afectados por el cáncer.
Efectos directos en el tejido.
Disfunción orgánica inducida por el cáncer u otros efectos tisulares.
Factores relacionados con el tratamiento
Radioterapia: dosis total, tamaño de la fracción, volumen del órgano o tejido expuesto.
Quimioterapia: tipo de fármaco, intensidad de las dosis, dosis acumulada, pauta de administración.
Recursos para la atención de apoyo al sobreviviente
Exámenes de detección según el riesgo
La American Society of Pediatric Hematology/Oncology, la International Society of Pediatric Oncology, la American Academy of Pediatrics, el Children’s Oncology Group (COG) y el Institute of Medicine reconocen la necesidad de establecer un seguimiento a largo plazo de los sobrevivientes de cáncer infantil. Se recomienda un seguimiento médico basado en el riesgo que incluya un plan sistemático para los exámenes de detección, la vigilancia y la prevención de por vida que incorpore cálculos de riesgo basados en los siguientes aspectos:[49]
Cáncer anterior.
Tratamiento del cáncer.
Predisposición genética.
Comportamientos propios del estilo de vida.
Afecciones comórbidas.
Sexo.
Parte del seguimiento a largo plazo también se enfoca en el control apropiado del avance educativo y vocacional. Es posible que los tratamientos específicos para el cáncer infantil, en especial los que afectan de manera directa las estructuras del sistema nervioso, generen deficiencias sensoriales, motrices y neurocognitivas con consecuencias adversas en el estado funcional, los logros educativos y las oportunidades vocacionales futuras. En apoyo de ello, en una investigación del CCSS se observó lo siguiente:[50]
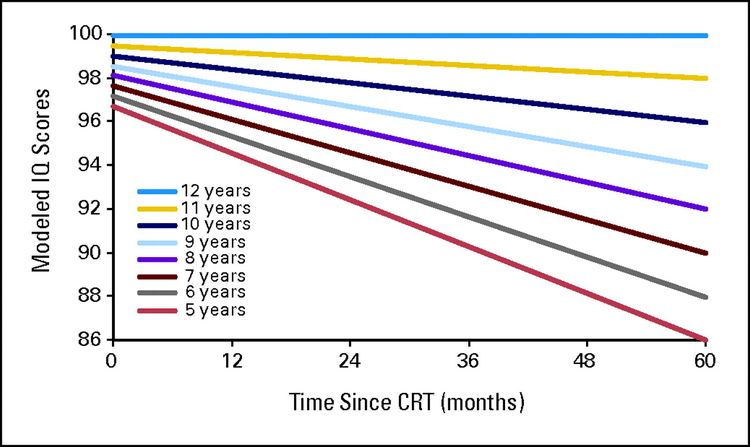
El tratamiento con dosis de radiación craneal de 25 Gy o más se vinculó con posibilidades más altas de desempleo (relacionado con la salud: OR, 3,47; intervalo de confianza [IC] 95 %, 2,54–4,74; búsqueda de trabajo: OR, 1,77; IC 95 %, 1,15–2,71).
Los sobrevivientes desempleados notificaron grados más altos de funcionamiento físico precario que los sobrevivientes empleados, un grado de escolaridad e ingresos más bajos, y mayor probabilidad de contar con seguro del sistema público, en comparación con los hermanos o hermanas desempleados.
Estos datos subrayan la importancia de facilitar el acceso de los sobrevivientes a los servicios educativos individualizados, los cuales revelaron una repercusión positiva en los logros educativos que,[51] a su vez, es posible que mejoren las oportunidades vocacionales.
Además de los exámenes de detección para los efectos tardíos médicos del tratamiento según el riesgo, también se hace hincapié en la repercusión de los comportamientos que afectan la salud en los riesgos relacionados con el cáncer. En los sobrevivientes del cáncer infantil se hace hincapié en los comportamientos que favorecen la salud. Las iniciativas educativas centradas en conductas para un estilo de vida saludable comprenden los siguientes aspectos:
Abstinencia del consumo de tabaco, el consumo excesivo de bebidas alcohólicas y el consumo de drogas ilícitas para reducir el riesgo de toxicidad orgánica y, posiblemente, neoplasias subsiguientes.
Prácticas alimentarias saludables (por ejemplo, una dieta rica en alimentos de origen vegetal y moderada en alimentos de origen animal) [52] y un estilo de vida activo para reducir las complicaciones metabólicas y cardiovasculares relacionadas con el tratamiento.
Actividad física regular para reducir los problemas neurocognitivos y mejorar los desenlaces psicológicos.[53,54]
Los sobrevivientes que realizaron actividad física constante, con el tiempo, tuvieron menos problemas neurocognitivos, como dificultades con la eficiencia de tareas, regulación emocional, organización y memoria. También experimentaron mayores mejoras neurocognitivas, en comparación con aquellos que tenían niveles de actividad irregulares.[53]
En los sobrevivientes, el ejercicio vigoroso se relacionó con una prevalencia más baja de depresión y somatización, así como menos deterioro en el funcionamiento físico, la salud general y la vitalidad, las limitaciones de la función emocional y los dominios de la calidad de vida de la salud mental.[54]
Es conveniente abordar de manera proactiva los comportamientos poco saludables y de riesgo porque varias investigaciones confirman que los sobrevivientes a largo plazo consumen tabaco y bebidas alcohólicas y llevan estilos de vida inactivos, a pesar del mayor riesgo de efectos tardíos cardíacos, pulmonares y metabólicos.[55-57]
Acceso de los sobrevivientes a la atención según el riesgo
La mayoría de los sobrevivientes de cáncer infantil no reciben la atención que se recomienda según el riesgo. En el CCSS se observó lo siguiente:
De los sobrevivientes, el 92,8 % notificó que recibió algún tipo de atención médica en el año anterior.[58]
Casi un 40 % notificó que recibió atención centrada en el cáncer previo (atención enfocada en el sobreviviente).[58]
La vigilancia de los casos nuevos de cáncer fue muy baja en sobrevivientes que presentaron el riesgo más alto de cáncer de colon, mama o piel; lo que indica que los sobrevivientes y sus médicos necesitan recibir educación sobre el riesgo de neoplasias subsiguientes y la vigilancia recomendada.[59]
Los factores sociodemográficos se han vinculado con la disminución de las tasas de atención de seguimiento a medida que pasa el tiempo desde el momento del diagnóstico. En la encuesta más reciente de seguimiento de los participantes del CCSS, los varones que tenían ingresos familiares anuales inferiores a $20 000 y menores logros educativos (educación de bachillerato como máximo) tuvieron mayor probabilidad de notificar no haber recibido atención para su salud. Esta tendencia es preocupante porque la prevalencia de las afecciones crónicas aumenta a medida que pasa más tiempo desde el diagnóstico de cáncer en los adultos tratados por cáncer durante la niñez.[60]
En un estudio en el que participaron 975 adultos sobrevivientes de cáncer infantil, se identificaron factores relacionados con la asistencia a las consultas médicas relacionadas con el cáncer recomendadas según el riesgo. El riesgo relativo de asistir a una consulta relacionada con el cáncer fue mayor en los sobrevivientes que tenían las siguientes características:[61]
Otorgaban más importancia a estas consultas.
Percibían una mayor susceptibilidad a presentar problemas de salud.
Padecían un problema de salud crónico relacionado con el cáncer de moderado a potencialmente mortal.
Estaban en contacto con personal de atención primaria por un problema relacionado con el cáncer.
Habían recibido un plan de atención para el tratamiento del cáncer.
Expresaban una mayor confianza en la capacidad de los médicos para responder preguntas e inquietudes.
El acceso al seguro médico parece desempeñar una función importante en la atención de los sobrevivientes según su riesgo.[62,63] La falta de acceso a un seguro médico afecta los siguientes aspectos:
Consultas relacionadas con el cáncer. En el CCSS, fue menos probable que los sobrevivientes sin seguro médico notificaran una consulta relacionada con el cáncer que quienes tenían un seguro privado (riesgo relativo ajustado, 0,83; IC 95 %, 0,75–0,91) o una consulta en un centro oncológico (riesgo relativo ajustado, 0,83; IC 95 %, 0,71–0,98). Los sobrevivientes sin seguro tenían índices más bajos de utilización en todas las mediciones de atención que los sobrevivientes con seguro privado. En contraste, fue más probable que los sobrevivientes con seguro público notificaran haber asistido a una consulta relacionada con el cáncer (riesgo relativo ajustado, 1,22; IC 95 %, 1,11–1,35) o una consulta en un centro oncológico (riesgo relativo ajustado 1,41; IC 95 %, 1,18–1,70) que los sobrevivientes con seguro privado.[62]
Desenlaces de atención de la salud. En un estudio, en el que se compararon los desenlaces de atención de la salud a largo plazo para los sobrevivientes de cáncer diagnosticado durante la juventud o adultez temprana con los desenlaces de personas sin antecedentes de cáncer, la proporción de sobrevivientes sin seguro médico no difirió entre los 2 grupos.[64]
Carga económica. Es posible que los subgrupos de adultos sobrevivientes de cáncer infantil tengan un riesgo adicional de enfrentar obstáculos para la atención de la salud debido a dificultades económicas. Los sobrevivientes más jóvenes (20–29 años de edad), las mujeres, los sobrevivientes que no eran blancos y los sobrevivientes que notificaron tener una salud más precaria enfrentaron más barreras económicas, lo que quizás impida la detección temprana de efectos tardíos.[64] Los sobrevivientes tienen más probabilidades que sus hermanos o hermanas de renunciar a la atención médica necesaria por causa de dificultades económicas.[65]
En términos generales, la falta de seguro médico sigue siendo una preocupación importante para los sobrevivientes de cáncer infantil debido a cuestiones de salud, desempleo y otros factores sociales.[66,67] La legislación, como la Ley de Portabilidad y Responsabilidad de Seguros Médicos (HIPAA),[68,69] mejoró el acceso y la retención del seguro médico de los sobrevivientes; sin embargo, no se han estudiado bien ni la calidad ni las limitaciones relacionadas con estas normas.
Transición a la atención del sobreviviente
Programas de seguimiento a largo plazo
La transición de la atención pediátrica al entorno de atención de salud para adultos es necesaria para la mayoría de los sobrevivientes de cáncer infantil en los Estados Unidos.
Cuando se dispone de programas multidisciplinarios de seguimiento a largo plazo en el centro oncológico pediátrico, se trabaja en colaboración con los médicos de la comunidad para proporcionar atención a los sobrevivientes de cáncer infantil. Este tipo de atención compartida se propuso como modelo óptimo para facilitar la coordinación entre el equipo del centro de oncología y los grupos médicos de la comunidad que atienden a los sobrevivientes.[70]
Un servicio esencial de los programas de seguimiento a largo plazo es la organización de un plan de atención individualizado de supervivencia que incluya los siguientes aspectos:
Detalles acerca de las intervenciones terapéuticas administradas para el cáncer infantil y sus riesgos para la salud (por ejemplo, tipo y dosis acumulada de quimioterapia, campos y dosis de radioterapia, procedimientos quirúrgicos, transfusiones de hemoderivados y trasplante de células hematopoyéticas).
Recomendaciones personalizadas para los exámenes médicos de detección.
Información acerca de los factores de estilo de vida que modifican los riesgos.
En una investigación del CCSS en la que se analizaron las percepciones sobre la salud futura y el riesgo de cáncer se resaltó la importancia de continuar la educación de los sobrevivientes durante las evaluaciones del seguimiento a largo plazo. En un subgrupo importante de adultos sobrevivientes se notificó falta de preocupación sobre los riesgos futuros de salud (24 %) y de cáncer subsiguiente (35 %), incluso después de la exposición a tratamientos relacionados con un aumento del riesgo. Estas conclusiones señalan el problema de que los sobrevivientes quizás sean menos propensos a participar en exámenes de detección beneficiosos y en actividades de reducción del riesgo.[71]
En el CCSS se evaluaron las prácticas de vigilancia y detección de 11 337 sobrevivientes de cáncer infantil. Se estableció que menos de la mitad de los sobrevivientes de riesgo alto que tenían la susceptibilidad más alta a presentar NMS o disfunción cardíaca recibieron la vigilancia recomendada, lo que acarrea riesgo de morbilidad y mortalidad prevenibles.[59]
Entre los sobrevivientes, el 27 % contaba con un plan de atención de seguimiento y, entre el personal de atención primaria, el 20 % tenía dicho plan. Los sobrevivientes tratados después de 1990 tenían más probabilidades de contar con un plan de atención del sobreviviente.
Los sobrevivientes de riesgo alto que tenían un plan de atención del sobreviviente exhibieron mayor cumplimiento con la vigilancia recomendada por el COG para la mama (22 vs. 8 %), la piel (35 vs. 23 %) y el corazón (67 vs. 33 %). La existencia de un plan de atención del sobreviviente se relacionó con mayor cumplimiento de la vigilancia de la piel (40 vs. 23 %).
Entre 2007 y 2016, en los sobrevivientes de riesgo alto, aumentó el cumplimiento de la vigilancia colorrectal (14 a 41 %, P < 0,001) y cardíaca (22 a 38 %, P < 0,001) y disminuyó el cumplimiento de la vigilancia mamaria (38 a 13 %, P < 0,001).
En el caso de los sobrevivientes de riesgo medio, se observó mejor cumplimiento de las recomendaciones expedidas por la American Cancer Society para los exámenes de detección mamarios (57 %), cervicouterinos (84 %) y colorrectales (69 %), en comparación con las recomendaciones expedidas por el COG. La existencia de un plan de atención del sobreviviente se relacionó con un mayor cumplimiento de los exámenes de detección mamarios y colorrectales. Los sobrevivientes cumplieron en menor medida con los exámenes de detección mamarios que la población general y también cumplieron en menor medida con los exámenes de detección cervicouterinos que sus hermanos o hermanas.
En el caso de los sobrevivientes que no recibieron esta información, el COG ofrece una plantilla, en inglés, que los sobrevivientes a veces usan para organizar un resumen del tratamiento personal. Para obtener más información en inglés, consultar las Survivorship Guidelines, Appendix 1 del COG.
Directrices del Children's Oncology Group para el seguimiento a largo plazo de sobrevivientes de cáncer diagnosticado durante la niñez, adolescencia o adultez temprana
Para facilitar el acceso del sobreviviente y el proveedor a información sucinta para guiar la atención según el riesgo, los investigadores del COG organizaron un compendio de recomendaciones de vigilancia sanitaria según la exposición y el riesgo con el fin de estandarizar la atención de los sobrevivientes de cáncer infantil.[72]
El compendio de recursos incluye lo siguiente:
Directrices de seguimiento a largo plazo. El documento en inglés del COG Long-Term Follow-Up Guidelines for Survivors of Childhood, Adolescent, and Young Adult Cancers contiene directrices apropiadas para sobrevivientes asintomáticos que se presentan para una evaluación médica de rutina a partir de las exposiciones que se realiza 2 o más años después de finalizar el tratamiento.
Health Links. Los materiales educativos para pacientes, conocidos como Health Links, proporcionan información detallada en inglés con directrices sobre temas específicos para mejorar el mantenimiento y la promoción de la salud en esta población de sobrevivientes de cáncer.[73]
La información sobre los efectos tardíos se sintetiza en los cuadros de este resumen.
Varios grupos se han dedicado a la investigación a fin de evaluar el rendimiento de los exámenes de detección según el riesgo, como lo recomiendan el COG y otros grupos cooperativos de oncología pediátrica.[7,74,75] Entre las consideraciones pertinentes a la interpretación de los resultados de estos estudios se incluyen las siguientes:
Variabilidad de la edad de la cohorte en el momento del tratamiento.
Edad en el momento del examen de detección.
Tiempo transcurrido desde el tratamiento del cáncer.
Sesgo de participación.
En conjunto, estos estudios demuestran que los exámenes de detección permiten identificar una proporción importante de personas con complicaciones clínicas de diferentes grados de gravedad relacionadas con el tratamiento que no se habían reconocido antes. En los resultados de los estudios también se identificaron evaluaciones de rendimiento bajo que alentaron revisiones de las recomendaciones sobre los exámenes de detección. En las investigaciones en curso se evalúa la rentabilidad de los exámenes de detección en un contexto en el que se toman en cuenta los beneficios, los riesgos y los perjuicios.
Bibliografía
National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.
Lorenzi MF, Xie L, Rogers PC, et al.: Hospital-related morbidity among childhood cancer survivors in British Columbia, Canada: report of the childhood, adolescent, young adult cancer survivors (CAYACS) program. Int J Cancer 128 (7): 1624-31, 2011. [PUBMED Abstract]
Rebholz CE, Reulen RC, Toogood AA, et al.: Health care use of long-term survivors of childhood cancer: the British Childhood Cancer Survivor Study. J Clin Oncol 29 (31): 4181-8, 2011. [PUBMED Abstract]
Armstrong GT, Kawashima T, Leisenring W, et al.: Aging and risk of severe, disabling, life-threatening, and fatal events in the childhood cancer survivor study. J Clin Oncol 32 (12): 1218-27, 2014. [PUBMED Abstract]
Teepen JC, Kok JL, Feijen EAM, et al.: Questionnaire- and linkage-based outcomes in Dutch childhood cancer survivors: Methodology of the DCCSS LATER study part 1. Cancer Med 12 (6): 7588-7602, 2023. [PUBMED Abstract]
Geenen MM, Cardous-Ubbink MC, Kremer LC, et al.: Medical assessment of adverse health outcomes in long-term survivors of childhood cancer. JAMA 297 (24): 2705-15, 2007. [PUBMED Abstract]
Hudson MM, Ness KK, Gurney JG, et al.: Clinical ascertainment of health outcomes among adults treated for childhood cancer. JAMA 309 (22): 2371-81, 2013. [PUBMED Abstract]
Feijen EAM, Teepen JC, van Dulmen-den Broeder E, et al.: Clinical evaluation of late outcomes in Dutch childhood cancer survivors: Methodology of the DCCSS LATER 2 study. Pediatr Blood Cancer 70 (5): e30212, 2023. [PUBMED Abstract]
Kurt BA, Nolan VG, Ness KK, et al.: Hospitalization rates among survivors of childhood cancer in the Childhood Cancer Survivor Study cohort. Pediatr Blood Cancer 59 (1): 126-32, 2012. [PUBMED Abstract]
Zhang Y, Lorenzi MF, Goddard K, et al.: Late morbidity leading to hospitalization among 5-year survivors of young adult cancer: a report of the childhood, adolescent and young adult cancer survivors research program. Int J Cancer 134 (5): 1174-82, 2014. [PUBMED Abstract]
Sørensen GV, Winther JF, de Fine Licht S, et al.: Long-Term Risk of Hospitalization Among Five-Year Survivors of Childhood Leukemia in the Nordic Countries. J Natl Cancer Inst 111 (9): 943-951, 2019. [PUBMED Abstract]
Emerson MA, Olshan AF, Chow EJ, et al.: Hospitalization and Mortality Outcomes Among Childhood Cancer Survivors by Race, Ethnicity, and Time Since Diagnosis. JAMA Netw Open 5 (6): e2219122, 2022. [PUBMED Abstract]
Streefkerk N, Tissing WJE, Korevaar JC, et al.: A detailed insight in the high risks of hospitalizations in long-term childhood cancer survivors-A Dutch LATER linkage study. PLoS One 15 (5): e0232708, 2020. [PUBMED Abstract]
Berbis J, Michel G, Chastagner P, et al.: A French cohort of childhood leukemia survivors: impact of hematopoietic stem cell transplantation on health status and quality of life. Biol Blood Marrow Transplant 19 (7): 1065-72, 2013. [PUBMED Abstract]
Phillips SM, Padgett LS, Leisenring WM, et al.: Survivors of childhood cancer in the United States: prevalence and burden of morbidity. Cancer Epidemiol Biomarkers Prev 24 (4): 653-63, 2015. [PUBMED Abstract]
Bhakta N, Liu Q, Ness KK, et al.: The cumulative burden of surviving childhood cancer: an initial report from the St Jude Lifetime Cohort Study (SJLIFE). Lancet 390 (10112): 2569-2582, 2017. [PUBMED Abstract]
Salloum R, Chen Y, Yasui Y, et al.: Late Morbidity and Mortality Among Medulloblastoma Survivors Diagnosed Across Three Decades: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 37 (9): 731-740, 2019. [PUBMED Abstract]
Streefkerk N, Teepen JC, Feijen EAM, et al.: The cumulative burden of self-reported, clinically relevant outcomes in long-term childhood cancer survivors and implications for survivorship care: A DCCSS LATER study. Cancer 130 (8): 1349-1358, 2024. [PUBMED Abstract]
Esbenshade AJ, Lu L, Friedman DL, et al.: Accumulation of Chronic Disease Among Survivors of Childhood Cancer Predicts Early Mortality. J Clin Oncol 41 (20): 3629-3641, 2023. [PUBMED Abstract]
Ehrhardt MJ, Williams AM, Liu Q, et al.: Cumulative burden of chronic health conditions among adolescent and young adult survivors of childhood cancer: Identification of vulnerable groups at key medical transitions. Pediatr Blood Cancer 68 (6): e29030, 2021. [PUBMED Abstract]
Vuotto SC, Krull KR, Li C, et al.: Impact of chronic disease on emotional distress in adult survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 123 (3): 521-528, 2017. [PUBMED Abstract]
Hudson MM, Oeffinger KC, Jones K, et al.: Age-dependent changes in health status in the Childhood Cancer Survivor cohort. J Clin Oncol 33 (5): 479-91, 2015. [PUBMED Abstract]
Liu Q, Leisenring WM, Ness KK, et al.: Racial/Ethnic Differences in Adverse Outcomes Among Childhood Cancer Survivors: The Childhood Cancer Survivor Study. J Clin Oncol 34 (14): 1634-43, 2016. [PUBMED Abstract]
Ehrhardt MJ, Bhakta N, Liu Q, et al.: Absence of Basal Cell Carcinoma in Irradiated Childhood Cancer Survivors of Black Race: A Report from the St. Jude Lifetime Cohort Study. Cancer Epidemiol Biomarkers Prev 25 (9): 1356-60, 2016. [PUBMED Abstract]
Turcotte LM, Liu Q, Yasui Y, et al.: Temporal Trends in Treatment and Subsequent Neoplasm Risk Among 5-Year Survivors of Childhood Cancer, 1970-2015. JAMA 317 (8): 814-824, 2017. [PUBMED Abstract]
Gibson TM, Mostoufi-Moab S, Stratton KL, et al.: Temporal patterns in the risk of chronic health conditions in survivors of childhood cancer diagnosed 1970-99: a report from the Childhood Cancer Survivor Study cohort. Lancet Oncol 19 (12): 1590-1601, 2018. [PUBMED Abstract]
Ness KK, Hudson MM, Jones KE, et al.: Effect of Temporal Changes in Therapeutic Exposure on Self-reported Health Status in Childhood Cancer Survivors. Ann Intern Med 166 (2): 89-98, 2017. [PUBMED Abstract]
Armstrong GT, Chen Y, Yasui Y, et al.: Reduction in Late Mortality among 5-Year Survivors of Childhood Cancer. N Engl J Med 374 (9): 833-42, 2016. [PUBMED Abstract]
Holmqvist AS, Chen Y, Wu J, et al.: Late mortality after autologous blood or marrow transplantation in childhood: a Blood or Marrow Transplant Survivor Study-2 report. Blood 131 (24): 2720-2729, 2018. [PUBMED Abstract]
Bagnasco F, Caruso S, Andreano A, et al.: Late mortality and causes of death among 5-year survivors of childhood cancer diagnosed in the period 1960-1999 and registered in the Italian Off-Therapy Registry. Eur J Cancer 110: 86-97, 2019. [PUBMED Abstract]
Byrne J, Schmidtmann I, Rashid H, et al.: Impact of era of diagnosis on cause-specific late mortality among 77 423 five-year European survivors of childhood and adolescent cancer: The PanCareSurFup consortium. Int J Cancer 150 (3): 406-419, 2022. [PUBMED Abstract]
Kilsdonk E, van Dulmen-den Broeder E, van Leeuwen FE, et al.: Late Mortality in Childhood Cancer Survivors according to Pediatric Cancer Diagnosis and Treatment Era in the Dutch LATER Cohort. Cancer Invest 40 (5): 413-424, 2022. [PUBMED Abstract]
Dixon SB, Liu Q, Chow EJ, et al.: Specific causes of excess late mortality and association with modifiable risk factors among survivors of childhood cancer: a report from the Childhood Cancer Survivor Study cohort. Lancet 401 (10386): 1447-1457, 2023. [PUBMED Abstract]
Mertens AC, Yong J, Dietz AC, et al.: Conditional survival in pediatric malignancies: analysis of data from the Childhood Cancer Survivor Study and the Surveillance, Epidemiology, and End Results Program. Cancer 121 (7): 1108-17, 2015. [PUBMED Abstract]
Fidler MM, Reulen RC, Winter DL, et al.: Long term cause specific mortality among 34 489 five year survivors of childhood cancer in Great Britain: population based cohort study. BMJ 354: i4351, 2016. [PUBMED Abstract]
Holmqvist AS, Chen Y, Wu J, et al.: Assessment of Late Mortality Risk After Allogeneic Blood or Marrow Transplantation Performed in Childhood. JAMA Oncol 4 (12): e182453, 2018. [PUBMED Abstract]
Dixon SB, Chen Y, Yasui Y, et al.: Reduced Morbidity and Mortality in Survivors of Childhood Acute Lymphoblastic Leukemia: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 38 (29): 3418-3429, 2020. [PUBMED Abstract]
Turcotte LM, Whitton JA, Leisenring WM, et al.: Chronic conditions, late mortality, and health status after childhood AML: a Childhood Cancer Survivor Study report. Blood 141 (1): 90-101, 2023. [PUBMED Abstract]
Ehrhardt MJ, Liu Q, Dixon SB, et al.: Association of Modifiable Health Conditions and Social Determinants of Health With Late Mortality in Survivors of Childhood Cancer. JAMA Netw Open 6 (2): e2255395, 2023. [PUBMED Abstract]
Chen C, Qin N, Wang M, et al.: Cancer germline predisposing variants and late mortality from subsequent malignant neoplasms among long-term childhood cancer survivors: a report from the St Jude Lifetime Cohort and the Childhood Cancer Survivor Study. Lancet Oncol 24 (10): 1147-1156, 2023. [PUBMED Abstract]
Anderson C, Smitherman AB, Nichols HB: Conditional relative survival among long-term survivors of adolescent and young adult cancers. Cancer 124 (14): 3037-3043, 2018. [PUBMED Abstract]
Anderson C, Nichols HB: Trends in Late Mortality Among Adolescent and Young Adult Cancer Survivors. J Natl Cancer Inst 112 (10): 994-1002, 2020. [PUBMED Abstract]
Suh E, Stratton KL, Leisenring WM, et al.: Late mortality and chronic health conditions in long-term survivors of early-adolescent and young adult cancers: a retrospective cohort analysis from the Childhood Cancer Survivor Study. Lancet Oncol 21 (3): 421-435, 2020. [PUBMED Abstract]
Rossetti S, Juul SJ, Eriksson F, et al.: Long-term cause-specific mortality in adolescent and young adult Hodgkin lymphoma patients treated with contemporary regimens-A nationwide Danish cohort study. Br J Haematol 205 (4): 1374-1382, 2024. [PUBMED Abstract]
Armenian SH, Xu L, Cannavale KL, et al.: Cause-specific mortality in survivors of adolescent and young adult cancer. Cancer 126 (10): 2305-2316, 2020. [PUBMED Abstract]
Chao C, Bhatia S, Xu L, et al.: Chronic Comorbidities Among Survivors of Adolescent and Young Adult Cancer. J Clin Oncol 38 (27): 3161-3174, 2020. [PUBMED Abstract]
Hudson MM, Armenian SH, Armstrong GT, et al.: Optimization of Health and Extension of Lifespan Through Childhood Cancer Survivorship Research. J Clin Oncol 36 (21): 2133-2134, 2018. [PUBMED Abstract]
Kremer LC, Mulder RL, Oeffinger KC, et al.: A worldwide collaboration to harmonize guidelines for the long-term follow-up of childhood and young adult cancer survivors: a report from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Pediatr Blood Cancer 60 (4): 543-9, 2013. [PUBMED Abstract]
Dixon SB, Bjornard KL, Alberts NM, et al.: Factors influencing risk-based care of the childhood cancer survivor in the 21st century. CA Cancer J Clin 68 (2): 133-152, 2018. [PUBMED Abstract]
Kirchhoff AC, Leisenring W, Krull KR, et al.: Unemployment among adult survivors of childhood cancer: a report from the childhood cancer survivor study. Med Care 48 (11): 1015-25, 2010. [PUBMED Abstract]
Mitby PA, Robison LL, Whitton JA, et al.: Utilization of special education services and educational attainment among long-term survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. Cancer 97 (4): 1115-26, 2003. [PUBMED Abstract]
Lan T, Wang M, Ehrhardt MJ, et al.: Adherence to healthy diet and risk of cardiovascular disease in adult survivors of childhood cancer in the St. Jude Lifetime Cohort: a cross-sectional study. BMC Med 21 (1): 242, 2023. [PUBMED Abstract]
Barlow-Krelina E, Chen Y, Yasui Y, et al.: Consistent Physical Activity and Future Neurocognitive Problems in Adult Survivors of Childhood Cancers: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 38 (18): 2041-2052, 2020. [PUBMED Abstract]
Tonorezos ES, Ford JS, Wang L, et al.: Impact of exercise on psychological burden in adult survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 125 (17): 3059-3067, 2019. [PUBMED Abstract]
Lown EA, Hijiya N, Zhang N, et al.: Patterns and predictors of clustered risky health behaviors among adult survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 122 (17): 2747-56, 2016. [PUBMED Abstract]
Gibson TM, Liu W, Armstrong GT, et al.: Longitudinal smoking patterns in survivors of childhood cancer: An update from the Childhood Cancer Survivor Study. Cancer 121 (22): 4035-43, 2015. [PUBMED Abstract]
Devine KA, Mertens AC, Whitton JA, et al.: Factors associated with physical activity among adolescent and young adult survivors of early childhood cancer: A report from the childhood cancer survivor study (CCSS). Psychooncology 27 (2): 613-619, 2018. [PUBMED Abstract]
Mueller EL, Park ER, Kirchhoff AC, et al.: Insurance, chronic health conditions, and utilization of primary and specialty outpatient services: a Childhood Cancer Survivor Study report. J Cancer Surviv 12 (5): 639-646, 2018. [PUBMED Abstract]
Yan AP, Chen Y, Henderson TO, et al.: Adherence to Surveillance for Second Malignant Neoplasms and Cardiac Dysfunction in Childhood Cancer Survivors: A Childhood Cancer Survivor Study. J Clin Oncol 38 (15): 1711-1722, 2020. [PUBMED Abstract]
Casillas J, Oeffinger KC, Hudson MM, et al.: Identifying Predictors of Longitudinal Decline in the Level of Medical Care Received by Adult Survivors of Childhood Cancer: A Report from the Childhood Cancer Survivor Study. Health Serv Res 50 (4): 1021-42, 2015. [PUBMED Abstract]
Ford JS, Tonorezos ES, Mertens AC, et al.: Barriers and facilitators of risk-based health care for adult survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 126 (3): 619-627, 2020. [PUBMED Abstract]
Casillas J, Castellino SM, Hudson MM, et al.: Impact of insurance type on survivor-focused and general preventive health care utilization in adult survivors of childhood cancer: the Childhood Cancer Survivor Study (CCSS). Cancer 117 (9): 1966-75, 2011. [PUBMED Abstract]
Keegan TH, Tao L, DeRouen MC, et al.: Medical care in adolescents and young adult cancer survivors: what are the biggest access-related barriers? J Cancer Surviv 8 (2): 282-92, 2014. [PUBMED Abstract]
Kirchhoff AC, Lyles CR, Fluchel M, et al.: Limitations in health care access and utilization among long-term survivors of adolescent and young adult cancer. Cancer 118 (23): 5964-72, 2012. [PUBMED Abstract]
Nathan PC, Huang IC, Chen Y, et al.: Financial Hardship in Adult Survivors of Childhood Cancer in the Era After Implementation of the Affordable Care Act: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 41 (5): 1000-1010, 2023. [PUBMED Abstract]
Kirchhoff AC, Kuhlthau K, Pajolek H, et al.: Employer-sponsored health insurance coverage limitations: results from the Childhood Cancer Survivor Study. Support Care Cancer 21 (2): 377-83, 2013. [PUBMED Abstract]
Kuhlthau KA, Nipp RD, Shui A, et al.: Health insurance coverage, care accessibility and affordability for adult survivors of childhood cancer: a cross-sectional study of a nationally representative database. J Cancer Surviv 10 (6): 964-971, 2016. [PUBMED Abstract]
Park ER, Kirchhoff AC, Zallen JP, et al.: Childhood Cancer Survivor Study participants' perceptions and knowledge of health insurance coverage: implications for the Affordable Care Act. J Cancer Surviv 6 (3): 251-9, 2012. [PUBMED Abstract]
Warner EL, Park ER, Stroup A, et al.: Childhood cancer survivors' familiarity with and opinions of the Patient Protection and Affordable Care Act. J Oncol Pract 9 (5): 246-50, 2013. [PUBMED Abstract]
Jacobs LA, Shulman LN: Follow-up care of cancer survivors: challenges and solutions. Lancet Oncol 18 (1): e19-e29, 2017. [PUBMED Abstract]
Gibson TM, Li C, Armstrong GT, et al.: Perceptions of future health and cancer risk in adult survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 124 (16): 3436-3444, 2018. [PUBMED Abstract]
Landier W, Bhatia S, Eshelman DA, et al.: Development of risk-based guidelines for pediatric cancer survivors: the Children's Oncology Group Long-Term Follow-Up Guidelines from the Children's Oncology Group Late Effects Committee and Nursing Discipline. J Clin Oncol 22 (24): 4979-90, 2004. [PUBMED Abstract]
Eshelman D, Landier W, Sweeney T, et al.: Facilitating care for childhood cancer survivors: integrating children's oncology group long-term follow-up guidelines and health links in clinical practice. J Pediatr Oncol Nurs 21 (5): 271-80, 2004 Sep-Oct. [PUBMED Abstract]
Landier W, Armenian SH, Lee J, et al.: Yield of screening for long-term complications using the children's oncology group long-term follow-up guidelines. J Clin Oncol 30 (35): 4401-8, 2012. [PUBMED Abstract]
Wasilewski-Masker K, Mertens AC, Patterson B, et al.: Severity of health conditions identified in a pediatric cancer survivor program. Pediatr Blood Cancer 54 (7): 976-82, 2010. [PUBMED Abstract]
Neoplasias subsiguientes
Las neoplasias subsiguientes (NS) se definen como neoplasias con características histológicas diferentes a la neoplasia primaria, que se presentan por lo menos 2 meses después de finalizar el tratamiento de la neoplasia maligna primaria; las NS pueden ser benignas o malignas. Los sobrevivientes de cáncer infantil tienen un aumento del riesgo de NS, de causa multifactorial, que varía de acuerdo con los siguientes factores:
Factores relacionados con el huésped (es decir, características genéticas, funcionamiento inmunitario, estado hormonal).
Tratamiento del cáncer primario.
Exposiciones ambientales.
Factores relacionados con el estilo de vida.
Las NS son la principal causa de mortalidad tardía sin recaída (razón de mortalidad estandarizada, 15,2; intervalo de confianza [IC] 95 %, 13,9–16,6).[1] En el Childhood Cancer Survivor Study (CCSS) se notificaron las siguientes tasas de incidencia acumuladas a 30 años:[2]
Todas las neoplasias subsiguientes: un 20,5 % (IC 95 %, 19,1–21,8 %).
Cáncer de piel no melanoma (CPNM): un 9,1 % (IC 95 %, 8,1–10,1 %).
NS con características histológicas malignas (excluidos los CPNM): un 7,9 % (IC 95 %, 7,2–8,5 %).
Meningioma: un 3,1 % (IC 95 %, 2,5–3,8 %).
Esto representa un aumento del riesgo de NS entre los sobrevivientes de cáncer que es 6 veces mayor que el de la población general.[2]
El exceso de riesgo de NS se ha descrito en varios estudios.[3,4]
Evidencia (exceso de riesgo de NS):
En un estudio poblacional, en el que se aprovecharon los datos de registro, se evaluaron los segundos tumores primarios tempranos que se presentaron dentro de los 5 años de un primer cáncer primario diagnosticado antes de los 15 años (1971–2010).[5]
En el 0,4 % de los sobrevivientes de cáncer se presentaron segundos tumores primarios tempranos, lo que representa un exceso de riesgo de 7 veces (razón de incidencia estandarizada [RIE], 7,7; IC 95 %, 6,7–8,9).
El exceso de riesgo fue más alto en los niños diagnosticados entre 1981 y 1990 (CIE, 9,5; IC 95 %, 7,1–12,5), en comparación con décadas anteriores o posteriores (CIE, 6,5–7,5).
Los síndromes de predisposición al cáncer fueron un factor de repercusión en el 21 % de los niños con segundos tumores primarios tempranos y se sospechó su presencia en otro 5 %.
En un estudio internacional de casos y controles se combinaron datos para un análisis del riesgo de meningioma después del tratamiento del cáncer infantil. En el estudio se evaluó la magnitud de la relación dosis-respuesta de radiación, los posibles modificadores del riesgo de radiación y la función de la quimioterapia.[6]
El aumento de la dosis de radiación se relacionó con un aumento del riesgo de meningioma (exceso de oportunidad relativa [OR]/Gy, 1,44; IC 95 %, 0,6–3,6), sin evidencia de desviación de la linealidad.
La exposición a dosis de radiación de 24 Gy o más se relacionó con una probabilidad 30 veces más alta de presentar un meningioma (OR, 33,7; IC 95 %, 14,1–80,3).
Los pacientes de 10 años o más en el momento del tratamiento tuvieron una relación significativamente más baja entre la dosis de radiación y la respuesta, en comparación con aquellos que recibieron tratamiento antes de los 10 años (exceso de OR/Gy, 0,57; IC 95 %, 0,18–1,91 vs. 2,20; IC 95 %, 0,87–6,31).
El tratamiento con metotrexato se relacionó con un aumento del riesgo de meningioma (OR, 3,43; IC 95 %, 1,56–7,57), pero sin evidencia de una relación entre la dosis y la respuesta o interacción con la dosis de radiación.
El riesgo de meningioma relacionado con la exposición a la radiación se mantuvo significativamente elevado 30 años después del tratamiento (exceso de OR/Gy, 3,76; IC 95 %, 0,77–29,15).
En una cohorte del CCSS se notificó cualquier NS nueva (incluso neoplasias malignas, CPNM, meningiomas benignos y otras neoplasias benignas) que se presentara después de los 40 años de edad.[3]
A la edad de 55 años, la incidencia acumulada de cualquier NS nueva fue del 34,6 %. La incidencia de NS malignas fue del 16,3 %.
En un análisis multivariante, el sexo femenino y la exposición a la radiación terapéutica se relacionaron con un aumento del riesgo de presentar NS.
Más aún, mediante un seguimiento prolongado, se estableció que las NS múltiples son comunes en los sobrevivientes de cáncer infantil a medida que envejecen.[7,8]
Las personas tratadas en las épocas de tratamientos más recientes presentaron una disminución del riesgo de NS (incluso neoplasias malignas subsiguientes, CPNM y meningiomas benignos), en comparación con aquellas que se trataron antes; si bien esto se relacionó con la disminución de la exposición a la radiación terapéutica,
Las personas tratadas en la década de 1990 todavía tienen un aumento de riesgo de NS, en comparación con la población general.
Los investigadores del CCSS evaluaron la morbilidad y la mortalidad relacionadas con el meningioma en 4221 participantes que recibieron radioterapia craneal.[9]
La incidencia acumulada de un meningioma subsiguiente a los 40 años de edad fue del 5,6 % en este grupo de pacientes; además, la incidencia no tuvo una meseta demostrable.
Los factores de riesgo de un meningioma subsiguiente fueron el sexo femenino (cociente de riesgos instantáneos [CRI], 1,7; IC 95 %, 1,2–2,3) y las dosis más altas de radiación craneal (CRI, 2,6; IC 95 %, 1,6–4,2 después de 30 Gy o más).
Entre los sobrevivientes que notificaron meningioma, el riesgo de secuelas neurológicas 5 años o más después del diagnóstico del cáncer primario aumentó en términos de convulsiones (CRI, 10,0; IC 95 %, 7,0–15,3); deficiencias auditivas, vestibulares, visuales y sensoriales (CRI, 2,3; IC 95 %, 1,3–4,0); disfunción neurológica focal (CRI, 4,9; IC 95 %, 3,2–7,5) y cefaleas graves (CRI, 3,2; IC 95 %, 1,9–5,4).
Al cabo de una mediana de seguimiento de 72 meses después de un diagnóstico de meningioma, el 13 % de los pacientes había muerto; 6 de las muertes se atribuyeron al meningioma.
Los investigadores del Dutch Childhood Oncology Group (DCOG)-LATER evaluaron la incidencia acumulada del meningioma y formularon modelos del exceso de riesgo relativo de meningiomas benignos en pacientes de cáncer infantil.[10]
Entre 5843 sobrevivientes de cáncer infantil (mediana de seguimiento, 23,3 años; intervalo, 5,0–52,2 años), 97 pacientes presentaron un meningioma benigno; 80 después de recibir un volumen total de radiación craneal y 14 después de recibir un volumen parcial de radiación craneal.
La incidencia acumulada a 40 años de meningioma después de cualquier radiación craneal fue de 12,4 % (IC 95 %, 9,8–15,2 %).
En comparación con las dosis de radiación craneal de 1 a 19 Gy, la ausencia de radiación craneal se relacionó con un riesgo bajo de meningioma (CRI, 0,04; IC 95 %, 0,01–0,15), sin embargo se observó un aumento del riesgo para dosis de 20 a 39 Gy (CRI, 1,66; IC 95 %, 0,83–3,33) y 40 Gy o más (CRI, 2,81; IC 95 %, 1,30–6,08).
Los sobrevivientes que se diagnosticaron antes de los 5 años de edad mostraron un aumento significativo del riesgo (CRI, 2,38; IC 95 %, 1,39–4,07), en comparación con los pacientes diagnosticados entre los 10 y los 17 años.
El volumen de radiación no se relacionó de manera significativa con un aumento del riesgo (CRI para el volumen total vs. el volumen parcial, 1,66; IC 95 %, 0,86–3,22) y los efectos de la dosis no variaron significativamente en función de la edad de exposición o el volumen de radiación.
El tratamiento con carboplatino se relacionó con el riesgo de meningioma (CRI, 3,55; IC 95 %, 1,62–7,78), pero no se observó ninguna respuesta según la dosis y los 9 casos expuestos habían recibido dosis altas de radiación craneal.
En un estudio de seguimiento de la cohorte DCOG-LATER, el 90 % de los sobrevivientes con meningioma consultaron por presentar síntomas en lugar de detectarse en consultorios o clínicas de efectos tardíos, y el 32 % de los sobrevivientes presentaron meningiomas síncronos.[11]
Los investigadores del CCSS evaluaron también la relación entre la quimioterapia y las neoplasias malignas subsiguientes (NMS) en sobrevivientes a largo plazo que no habían sido irradiados.[12]
De 1498 NMS en 1344 sobrevivientes, 229 se presentaron en 206 sobrevivientes que solo recibieron quimioterapia.
La incidencia acumulada a 30 años de NMS fue del 3,9 % en el grupo de quimioterapia sola, el 9,0 % en el grupo de quimioterapia y radiación, el 10,8 % en el de radiación sola y el 3,4 % en el grupo que no recibió ninguno de los 2 tratamientos.
El razón de incidencia estandarizada (RIE) de las NMS aumentó para cualquier NMS (RIE, 2,8), leucemia o linfoma subsiguiente (RIE, 1,9), cáncer de mama (RIE, 4,6), sarcoma de tejido blando (RIE, 3,4), cáncer de tiroides (RIE, 3,8) y melanoma (RIE, 2,3).
La tasa de NMS se relacionó de manera significativa con la exposición a quimioterapia con derivados del platino en dosis superiores a 750 mg/m2 (tasa relativa, 2,7) y con alquilantes (tasa relativa, 1,2 por 5000 mg/m2).
La tasa de cáncer de mama exhibió una respuesta lineal en relación con la dosis (tasa relativa, 1,3 por 100 mg/m2) de exposición a una antraciclina.
Los investigadores del DCOG-LATER evaluaron la contribución de la quimioterapia al riesgo de un cáncer sólido en una cohorte grande de sobrevivientes de cáncer infantil diagnosticados entre 1963 y 2001 (mediana de seguimiento, 20,7 años).[13]
La incidencia acumulada de NS a 25 años fue del 3,9 % y no cambió a lo largo de las décadas.
Los sobrevivientes tratados con doxorrubicina exhibieron un aumento del riesgo relacionado con la dosis, de todos los cánceres sólidos y el cáncer de mama. Esta relación fue más fuerte en sobrevivientes de cánceres infantiles relacionados con el síndrome de Li-Fraumeni (leucemia, cáncer del sistema nervioso central [SNC] y sarcomas diferentes al de Ewing) que en sobrevivientes de otros cánceres.
Entre las mujeres sobrevivientes que no recibieron radiación torácica ni irradiación corporal total (ICT) y presentaron cáncer de mama (n = 31), los CRI para los terciles de dosis de doxorrubicina fueron de 1,3 (IC 95 %, 0,3–6,1), 5,6 (IC 95 %, 1,9–16,2) y 9,9 (IC 95 %, 4,2–23,8).
Se confirmó una relación entre la dosis de ciclofosfamida y la respuesta para un sarcoma subsiguiente; en particular, el sarcoma óseo. El CRI para el sarcoma subsiguiente fue de 3,1 (IC 95 %, 1,5–6,0) en los sobrevivientes que recibieron dosis de ciclofosfamida superiores a 9400 mg/m2 y de 2,6 (IC 95 %, 1,3–5,2) en aquellos que recibieron ifosfamida.
Los investigadores del estudio de cohorte St. Jude Life (SJLIFE) evaluaron la contribución al riesgo de NS de las variantes patogénicas y posiblemente patogénicas de genes de predisposición al cáncer en sobrevivientes de cáncer infantil.[14]
De los 3006 participantes del estudio en quienes se evaluó la secuenciación del genoma completo (30 veces), se diagnosticaron 1120 NS en 439 sobrevivientes (14,6 %) y se identificaron 175 variantes patogénicas y posiblemente patogénicas en el 5,8 % de los sobrevivientes; la prevalencia de variantes patogénicas y posiblemente patogénicas en sobrevivientes no irradiados con NS fue mucho más alta, del 18 %.
Las variantes se relacionaron con un incremento significativo en las tasas de cáncer de mama (riesgo relativo [RR], 13,9) y sarcoma (RR, 10,6) en los sobrevivientes irradiados y que presentaron cualquier NS (RR, 4,7), cáncer de mama (RR, 7,7), cáncer de piel no melanoma (RR, 11), y 2 o más NS de tipo histológico diferente (RR, 18,6).
Los portadores de variantes no presentaron aumento de la tasa de meningioma o cáncer de tiroides.
La incidencia y el tipo de NS dependen de los siguientes aspectos:
Diagnóstico del cáncer primario.
Tipo de tratamiento recibido.
Presencia de afecciones genéticas.
Las relaciones distintivas con exposiciones terapéuticas específicas condujeron a la clasificación de las NS en los siguientes 2 grupos diferentes:
Síndrome mielodisplásico posterior a tratamiento citotóxico (SMD-pCT) y leucemia mieloide aguda posterior a tratamiento citotóxico (LMA-pCT) (antes conocidos como SMD-t y LMA-t).
NS sólidas relacionadas con el tratamiento.
Síndrome mielodisplásico y leucemia mieloide aguda posteriores a tratamiento citotóxico
Se notificaron leucemias primarias subsiguientes en sobrevivientes de linfoma de Hodgkin, leucemia, sarcoma, tumores del SNC, linfoma no Hodgkin, neuroblastoma y tumor de Wilms. En una cohorte de casi 70 000 sobrevivientes a 5 años de cáncer infantil, estos presentaron un riesgo 4 veces más alto (CIE, 3,7) de leucemia, con un exceso de riesgo absoluto de 7,5. En particular, se notificó un riesgo relativo 6 veces más alto de presentar leucemia mieloide (CIE, 5,8).[15]
El riesgo se mantuvo elevado de manera significativa 20 años después de la neoplasia maligna primaria inicial (CIE, 2,4).
Los sobrevivientes de linfoma de Hodgkin presentaron el exceso de riesgo más alto de leucemias subsiguientes (CIE, 7,9) y, en particular, de leucemia mieloide (CIE, 12,1); el mayor exceso de riesgo se observó antes de los 20 años de seguimiento.
En un análisis agrupado se examinaron todos los estudios publicados con datos detallados sobre el tratamiento de niños con cáncer diagnosticado entre 1930 y 2000. Los datos de tratamiento incluyeron la dosis estimada de radiación dirigida a la médula ósea activa y la dosis de fármacos quimioterapéuticos específicos. En este informe, 147 casos de una segunda leucemia primaria (69 %, LMA) se emparejaron con 522 controles.[16]
Después de controlar por otros tratamientos, el uso de un inhibidor de la topoisomerasa II se relacionó con un aumento del riesgo de una segunda LMA (sin radiación y >2500 mg/m2 vs. ninguno de estos: OR, 14,3; IC 95 %, 2,7–75,1).
La dosis de radiación dirigida a la médula ósea activa también se relacionó con un aumento del riesgo de una segunda LMA entre los pacientes que no recibieron quimioterapia (>0–12 Gy: OR, 4,9; IC 95 %, 0,95–25,6), pero no entre aquellos que recibieron quimioterapia.
Fue más probable que una segunda leucemia ocurriera durante los 10 años posteriores al tratamiento del cáncer.[17]
Las características del SMD-pCT y la LMA-pCT son las siguientes:[18,19]
Latencia corta (<10 años desde el diagnóstico del cáncer primario). El riesgo del SMD-pCT y la LMA-pCT se estabiliza luego de 10 a 15 años. Si bien el riesgo de leucemia subsiguiente permanece muy elevado más allá de los 15 años posteriores al diagnóstico primario (CIE, 3,5; IC 95 %, 1,9–6,0), estos casos son relativamente poco frecuentes, con un exceso de riesgo absoluto de 0,02 casos por 1000 años-persona.[19]
Relación con los alquilantes y los inhibidores de la topoisomerasa II.
A partir de las definiciones actualizadas de la Organización Mundial de la Salud, el SMD-pCT y la LMA-pCT son trastornos clonales que se presentan en pacientes expuestos previamente a terapia citotóxica, ya sea quimioterapia o radioterapia de campo amplio, para una neoplasia no relacionada.[20] Los siguientes 2 tipos de SMD-pCT y LMA-pCT son los que se observan con mayor frecuencia en los sobrevivientes:
Tipo vinculado a un alquilante: los alquilantes relacionados con el SMD-pCT y la LMA-pCT son la ciclofosfamida, la ifosfamida, la clormetina (mecloretamina), el melfalán, el busulfano, las nitrosoureas, el clorambucilo y la dacarbazina.[21]
El riesgo de SMD o LMA relacionado con un alquilante depende de la dosis, con una latencia de 3 a 5 años después de la exposición; se vincula con anomalías que afectan los cromosomas 5 (-5/del(5q)) y 7 (-7/del(7q)).[21]
Tipo vinculado a un inhibidor de la topoisomerasa II: los inhibidores de la topoisomerasa II son el etopósido, el tenipósido y los fármacos relacionados con las antraciclinas.
La mayoría de las translocaciones que se observan en pacientes expuestos a inhibidores de la topoisomerasa II afectan la región de ruptura situada entre los exones 5 y 11 de la banda 11q23, y fusionan el KMT2A con un gen compañero.[21] La LMA-t vinculada a un inhibidor de la topoisomerasa II se presenta como una leucemia evidente después de un periodo de latencia de 6 meses a 3 años y se relaciona con translocaciones equilibradas que comprometen las bandas cromosómicas 11q23 o 21q22.[22]
Neoplasias subsiguientes sólidas relacionadas con el tratamiento
Las neoplasias subsiguientes (NS) sólidas relacionadas con el tratamiento representan el 80 % de todas las NS y exhiben una relación fuerte con la exposición a la radiación; asimismo, se caracterizan por una latencia que excede los 10 años. El riesgo de NS sólidas continúa en aumento a medida que se prolonga el seguimiento. El riesgo de NS sólidas es más alto en los siguientes casos:[4,13]
Edad temprana en el momento de la exposición a la radiación.
Dosis total alta de radiación.
Mayor período de seguimiento después de la exposición a la radiación.
Los subtipos histológicos de NS sólidas abarcan un conjunto de neoplasias que fluctúa entre lesiones benignas y malignas de grado bajo (por ejemplo, CPNM y meningiomas) y neoplasias malignas de grado alto (por ejemplo, cánceres de mama y glioblastomas) (consultar la Figura 4).[4,13,23,24]
Con un seguimiento más prolongado de las cohortes de adultos sobrevivientes de cáncer infantil, se han observado neoplasias epiteliales en los siguientes sitios:[13,29]
También se han observado NS benignas y de grado bajo, como los CPNM y los meningiomas, con un aumento de la prevalencia en los sobrevivientes tratados con radioterapia por un cáncer infantil.[4,10,30]
Neoplasias subsiguientes después de un trasplante de células madre hematopoyéticas
Los receptores de TCMH se tratan con quimioterapia de dosis alta y, con frecuencia, ICT, con lo cual su riesgo de NS es único en la población oncológica general.
En 4318 receptores de TCMH alogénico tratados por primera vez por LMA y leucemia mieloide crónica entre 1986 y 2005, y acondicionados con dosis altas de busulfano y ciclofosfamida (Bu-Cy), se notificaron 66 cánceres sólidos al cabo de una mediana de 6 años después del TCMH.[31]
La incidencia acumulada de cánceres sólidos nuevos fue del 0,9 % a los 5 años y del 2,4 % a los 10 años y es similar, con independencia de la exposición a la radiación.
El acondicionamiento con Bu-Cy sin ICT se relacionó con riesgos más altos de NS sólidas que los de la población general.
La enfermedad de injerto contra huésped crónica aumentó el riesgo de NS, sobre todo el de neoplasias que comprometen la cavidad oral.
En un estudio de 4905 sobrevivientes a 1 año de un trasplante de células hematopoyéticas (TCH) de tipo alogénico que se sometieron al trasplante entre 1969 y 2014 por enfermedades malignas o de otro tipo, y que se observaron durante una mediana de 12,5 años, se demostró un fuerte efecto de la dosis de ICT y la dosis de fraccionamiento en el riesgo de NS.[32]
La incidencia acumulada a 20 años de NS después de un TCMH en las personas tratadas antes de los 20 años de edad fue del 8,1 %.
El riesgo más alto de NS correspondió a los sobrevivientes expuestos a dosis altas de ICT de fracción única (6–12 Gy) o dosis muy altas de ICT fraccionadas (14,4–17,5 Gy).
Con dosis bajas de ICT (2–4,5 Gy), el riesgo de NS fue comparable al riesgo con quimioterapia sola, aunque era aún 2 veces más alto que el de la población general.
Entre las personas tratadas antes de los 20 años de edad, el número de NS fue 12,5 veces más alto que lo esperado para la población general, y el exceso de riesgo absoluto fue de 10,6 por 1000 años-persona. Los sobrevivientes tratados con TCMH a esta edad temprana fueron más propensos a presentar NS que los sobrevivientes que se trataron después de los 50 años de edad (CRI, 2,3).
Algunas de las NS sólidas bien establecidas se describen en las secciones a continuación:
Cáncer de mama
Las mujeres sobrevivientes de un cáncer diagnosticado durante la niñez, adolescencia y adultez temprana que recibieron radioterapia en campos que comprenden el tórax tienen un riesgo mayor de presentar cáncer de mama.
Las tasas de incidencia acumulada de cáncer de mama oscilan del 8 % al 20 % entre los 40 y los 45 años de edad en las sobrevivientes de cáncer infantil, y llegan al 35 % a los 50 años de edad en las sobrevivientes de linfoma de Hodgkin (tasas comparables a las observadas entre las portadoras de una variante del gen BRCA).[18,33-36]
La dosis de radiación y el volumen de la mama expuesta son factores importantes que inciden en el riesgo; los fármacos quimioterapéuticos específicos, en especial los alquilantes y las antraciclinas, también afectan el riesgo.[33,35,37]
Evidencia (exceso de riesgo de cáncer de mama):
El cáncer de mama es la NS sólida más común relacionada con el tratamiento después de un diagnóstico previo de linfoma de Hodgkin.[18,34] Entre las mujeres sobrevivientes de linfoma de Hodgkin infantil se ha observado lo siguiente:
Se notificó un exceso de riesgo de cáncer de mama en mujeres sobrevivientes tratadas con dosis altas y volumen ampliado de radiación a los 30 años de edad o antes.[38]
Los datos indican que las mujeres tratadas con dosis bajas de radiación dirigida al campo afectado también exhiben un exceso de riesgo de cáncer de mama.[39]
Las pacientes que recibieron un volumen limitado de radioterapia supradiafragmática (excluida la axila) tuvieron un riesgo significativamente más bajo de cánceres de mama subsiguientes que las pacientes sometidas a radioterapia dirigida al campo del manto.[37]
Para las pacientes tratadas con radioterapia dirigida al tórax antes de los 16 años, la incidencia acumulada de cáncer de mama a los 45 años de edad se acerca al 20 %.[18]
El período de latencia después de la irradiación dirigida al tórax oscila entre 8 y 10 años; el riesgo de cáncer de mama subsiguiente aumenta de manera lineal con la dosis de radiación (P para la tendencia < 0,001).[40]
El tratamiento con dosis acumuladas más altas de alquilantes y radiación ovárica de 5 Gy o más (exposiciones que predisponen a una menopausia prematura) se correlaciona con reducción del riesgo de cáncer de mama, lo que resalta la posible contribución de la estimulación hormonal a la carcinogénesis en la mama.[37,41,42]
El número observado de casos de cáncer de mama invasivo en sobrevivientes jóvenes (de <30 años), en comparación con lo que se espera en la población general, ha disminuido en las épocas de tratamiento recientes (1970, CIE, 55,0 vs. 1990, CIE, 14,3).[36]
Sin embargo, el riesgo de cáncer de mama también aumentó según los siguientes estudios en los que se expuso el tejido de la mama a dosis de radiación más bajas administradas para tratar un cáncer metastásico en el tórax o los pulmones (por ejemplo, tumor de Wilms o sarcoma).
En 116 niños de la cohorte del CCSS que recibieron tratamiento con 2 a 20 Gy dirigida a los pulmones (mediana de 14 Gy), el CIE para el cáncer de mama fue de 43,6 (IC 95 %, 27,1–70,1).[35]
En un informe de 2492 mujeres que participaron en los 4 primeros estudios de los National Wilms Tumor Studies (1969–1995) se abordó el exceso de riesgo de cáncer de mama.[43]
De las 369 mujeres que recibieron irradiación dirigida al tórax por un tumor de Wilms metastásico, 16 presentaron cáncer de mama invasivo (riesgo acumulado a los 40 años de edad, 14,8 %, [IC 95 %, 8,7–24,5 %]). El CIE de 27,6 (IC 95 %, 16,1–44,2) se determinó sobre la base de un seguimiento de 5010 años-persona.
De las 369 pacientes, las dosis de radiación dirigidas al tórax fueron de menos de 12 Gy en el 4 % de las pacientes, de 12 Gy en el 64 %, de 13 a 15 Gy en el 19 % y de más de 15 Gy en el 13 %.
Para todas las pacientes que presentaron cáncer de mama (con irradiación dirigida al tórax o sin esta), la mediana de edad en el momento del primer diagnóstico de cáncer de mama fue de 34,3 años (intervalo, 15,5–48,4) y la mediana de tiempo transcurrido desde el diagnóstico del tumor de Wilms fue de 27,1 años (intervalo, 7,9–35,7).
En un estudio colaborativo internacional se obtuvieron los datos individuales de 17 903 sobrevivientes de cáncer infantil. En el estudio se evaluaron los efectos dependientes de la dosis de las antraciclinas en la formación de cánceres de mama subsiguientes y las interacciones con la radioterapia dirigida al tórax. Hubo 782 sobrevivientes (4,4 %) que presentaron un cáncer de mama subsiguiente.[44]
La doxorrubicina se relacionó con un aumento dependiente de la dosis del riesgo de cáncer de mama subsiguiente (CRI cada 100 mg/m2, 1,24; IC 95 %, 1,18–1,31).
El riesgo aumento más del doble en los sobrevivientes que se trataron con dosis acumuladas de doxorrubicina de 200 mg/m2 o más, en comparación con aquellos que no recibieron doxorrubicina (CRI, 2,50 con 200–299 mg/m2; CRI, 2,33 con 300–399 mg/m2; CRI, 2,78 con ≥400 mg/m2).
Las relaciones con daunorrubicina no fueron estadísticamente significativas, mientras que la epirrubicina se relacionó con un mayor riesgo subsiguiente de cáncer de mama (exposición vs. ausencia de exposición: CRI, 3,25; IC 95 %, 1,59–6,63).
Los CRI cada 100 mg/m2 de doxorrubicina fueron de 1,11 (IC 95 %, 1,02–1,21) en los pacientes que recibieron radioterapia dirigida al tórax y de 1,26 (IC 95 %, 1,17–1,36) en aquellos que no recibieron radioterapia dirigida al tórax.
En el CCSS se evaluó el riesgo de presentar cáncer de mama después de la radioterapia y la quimioterapia con antraciclinas. En un estudio de casos y controles anidado de 271 sobrevivientes de cáncer infantil (diagnosticadas entre 1970 y 1986) que fueron luego diagnosticadas con cáncer de mama, la combinación de antraciclinas y radioterapia en la mama guardó relación con mayor riesgo de cáncer de mama, lo cual es congruente con una interacción aditiva.[33]
Para el grupo de estudio, la mediana de edad en el momento del diagnóstico del primer cáncer fue de 15 años y la mediana de edad en el momento del diagnóstico de cáncer de mama fue de 39 años.
La OR para el cáncer de mama aumentó con la dosis creciente de radiación en la mama (OR por cada 10 Gy, 3,9; IC 95 %, 2,5–6,5) y fue similar en los cánceres positivos para el receptor de estrógeno y en los negativos para este receptor.
La OR por cada 10 Gy en la mama fue superior en las mujeres que recibieron en los ovarios dosis de menos de 1 Gy (OR, 6,8; IC 95 %, 3,9–12,5) que en las mujeres que recibieron dosis de 15 Gy o más (OR, 1,4; IC 95 %, 1,0–6,4).
La OR para el cáncer de mama aumentó con la dosis acumulativa de antraciclina (OR por 100 mg/m2, 1,23; IC 95 %, 1,09–1,39; P < 0,01 para la tendencia).
Hubo una interacción aditiva entre la radioterapia y el tratamiento con antraciclinas. La OR fue de 19,1 (IC 95 %, 7,6–48,0) para la asociación combinada de tratamiento con antraciclinas y dosis de radiación dirigida a la mama de 10 Gy o más (en comparación con 0 a menos de 1 Gy) versus 9,6 (IC 95 %, 4,4–20,7) sin tratamiento con antraciclinas.
Las sobrevivientes de cáncer infantil que no estuvieron expuestas a radiación dirigida a la mama también tuvieron un aumento de riesgo de cáncer de mama a una edad temprana.
En el estudio SJLIFE se evaluó el riesgo subsiguiente de cáncer de mama en 1467 mujeres sobrevivientes de cáncer, así como el riesgo relacionado con la exposición a las antraciclinas. En el estudio también se evaluó si las imágenes de vigilancia afectan los desenlaces del cáncer de mama.[25]
En las mujeres que no recibieron radiación torácica ni antraciclinas, la incidencia acumulada de cáncer de mama fue del 2 % a los 35 años y del 15 % a los 50 años. En las mujeres tratadas con 250 mg/m2 o más de antraciclinas, las tasas fueron del 7 % a los 35 años de edad y del 46 % a los 50 años.
Las dosis de antraciclina de 250 mg/m2 o más siguieron presentando una asociación significativa con el aumento de riesgo de cáncer de mama en los modelos, excepto para las sobrevivientes con variantes génicas de predisposición al cáncer, las que recibían radiación dirigida al tórax de 10 Gy o más, o quienes presentaron ambas características.
En comparación con los cánceres de mama detectados por hallazgos físicos, fue más probable que los cánceres de mama detectados mediante imágenes o mastectomía profiláctica fueran carcinomas in situ, de masas más pequeñas, sin compromiso de los ganglios linfáticos y que se trataran sin quimioterapia.
Las imágenes duales con mamografía e imágenes por resonancia magnética (IRM) de la mama en esta cohorte conformaron un abordaje sensible y específico para identificar cánceres de mama que necesitan tratamiento menos intensivo que los cánceres de mama detectados mediante hallazgos físicos.
En una investigación del CCSS se examinó el riesgo de cáncer de mama en 3768 participantes mujeres que no recibieron radiación torácica.[45]
Se observó un exceso de riesgo 4 veces superior (CIE, 4,0; IC 95 %, 3,0–5,3) de cáncer de mama, en comparación con tasas en la población general.
El riesgo de cáncer de mama fue más alto en las sobrevivientes de sarcoma (CIE, 5,3; IC 95 %, 3,6–7,8) y leucemia (CIE, 4,1; IC 95 %, 2,4–6,9), cuya incidencia acumulada de cáncer de mama fue del 5,8 % y del 6,3 %, respectivamente, a los 45 años de edad.
El tratamiento con alquilantes y antraciclinas aumentó el riesgo de cáncer de mama en una forma dependiente de la dosis.
Los investigadores del CCSS también examinaron el riesgo de NS entre 7448 participantes que solo recibieron tratamiento con quimioterapia.[12]
La incidencia de cáncer de mama fue 4,6 veces mayor de lo que se esperaría en la población general (CIE, 4,6; IC 95 %, 3,5–6,0).
Se demostró una respuesta lineal en relación a la dosis entre las antraciclinas y la tasa de cáncer de mama (RR, 1,3/100 mg/m2; IC 95 %, 1,2–1,6).
Investigadores del DCOG-LATER evaluaron la contribución de la quimioterapia al riesgo de cáncer sólido en una cohorte grande de sobrevivientes de cáncer infantil diagnosticados entre 1963 y 2001.[13]
Los sobrevivientes tratados con doxorrubicina exhibieron un aumento del riesgo de cáncer de mama dependiente de la dosis (CRI, 3,1; IC 95 %, 1,4–6,5 en los sobrevivientes tratados con dosis de antraciclina de 250 mg/m2 o más).
La respuesta del cáncer de mama a la dosis de doxorrubicina fue más fuerte en sobrevivientes de cánceres relacionados con el síndrome de Li-Fraumeni (leucemia, SNC y sarcomas diferentes al de Ewing) que en sobrevivientes de otros cánceres.
En un estudio con datos del SEER Program se evaluaron las tendencias, entre 1990 y 2016, en el tratamiento quirúrgico y la incidencia acumulada a 5 y 10 años de cáncer de mama contralateral en mujeres tratadas con radioterapia por linfoma de Hodgkin antes de los 30 años y diagnosticadas con un cáncer de mama subsiguiente.[46]
En general, 263 (89,2 %) mujeres presentaron cáncer de mama unilateral, 50 (19,0 %) se sometieron a cirugía de conservación de la mama y 213 (81,0 %) se sometieron a mastectomía (40,5 % bilateral).
La incidencia a 5 años de cáncer de mama contralateral en mujeres sometidas a cirugía unilateral fue del 9,4 % (IC 95 %, 5,6–15,4 %), y aumentó al 20,2 % (IC 95 %, 13,7–29,2 %) a los 10 años y al 29,9 % (IC 95 %, 20,8–41,9 %) a los 15 años. Este hallazgo destaca la importancia de la vigilancia continua del cáncer de mama y la consideración de la mastectomía profiláctica.
El riesgo de cáncer de mama varía entre los sobrevivientes de cáncer infantil tratados con radioterapia dirigida al tórax y el riesgo se basa en múltiples factores clínicos. El primer modelo personalizado de predicción del riesgo de cáncer de mama se desarrolló y se validó usando cohortes multinacionales de mujeres sobrevivientes de cáncer a 5 años diagnosticadas antes de los 21 años y que recibieron irradiación torácica (n = 2147). El modelo incluye la edad actual, el campo de irradiación torácica, si la radioterapia torácica se administró en el año siguiente a la menarquia, la exposición a antraciclinas, la edad de la menopausia y el antecedente de cáncer de mama en un familiar de primer grado. El modelo está disponible en inglés como calculadora de riesgos en línea.[47]
Cáncer de mama subsiguiente versus de nueva aparición
En varios estudios se investigaron las características clínicas de cánceres de mama subsiguientes que surgen en mujeres que recibieron radioterapia como tratamiento de un cáncer infantil.[48-52]
En un estudio poblacional, se notificó que el cáncer de mama inducido por radiación tiene características clinicopatológicas más adversas, como se comprobó por un riesgo 2 veces mayor de cáncer de mama negativo para receptores de estrógeno y progesterona en las sobrevivientes de linfoma de Hodgkin después de 15 años, en comparación con las mujeres con cáncer de mama esporádico.[48]
En otros estudios se observó una proporción más alta de subtipos histológicos más malignos (por ejemplo, cáncer de mama triple negativo) que de cánceres invasivos esporádicos ajustados por edad.[49,50]
Estos hallazgos contradicen otros estudios de casos y controles más pequeños, realizados en hospitales, sobre cáncer de mama en sobrevivientes de linfoma de Hodgkin en los que no se identificó una variación significativa en el estado de receptores hormonales cuando se los comparó con los controles de cáncer de mama primario. En estudios previos tampoco se demostró una diferencia significativa en el riesgo general de los tumores de grado alto versus los de grado bajo.[51,52]
En un estudio con datos del SEER Program se evaluaron las características clinicopatológicas de 321 cánceres de mama diagnosticados en 257 mujeres (mediana de edad, 22 años; intervalo, 18–26 años en el momento del diagnóstico de linfoma de Hodgkin) que se trataron previamente con radiación para linfoma de Hodgkin.[53]
Las mujeres que presentaron cáncer de mama después de la radiación para linfoma de Hodgkin eran más jóvenes que las mujeres diagnosticadas con cáncer de mama sin neoplasia maligna previa (43 vs. 60 años) y tenían menos probabilidad de presentar carcinoma ductal in situ (0,4 vs. 14,9 %).
La radiación no se relacionó con características biológicas más precarias, en comparación con los cánceres de mama diagnosticados en mujeres de edad similar que no presentaban neoplasia maligna previa. Sin embargo, se encontraron proporciones más altas de tumores en el cuadrante superior izquierdo y tumores más pequeños (≤2 cm).
La mayoría de los tumores en los sobrevivientes de linfoma de Hodgkin eran sensibles a las hormonas (casi 2 tercios eran positivos para receptores de estrógeno y negativos para HER2). Este hallazgo indica que la prevención endocrina puede ser una opción para esta población de riesgo alto.
Mortalidad después del cáncer de mama subsiguiente
En un estudio de mujeres participantes en el CCSS, que luego recibieron un diagnóstico de cáncer de mama (n = 274) y se emparejaron con un grupo control de mujeres (n = 1095) con cáncer de mama de nueva aparición, se observó lo siguiente:[54]
Las sobrevivientes de cáncer infantil tenían tasas de mortalidad elevadas (CRI, 2,2; IC 95 %, 1,7–3,0), incluso después de ajustar el tratamiento del cáncer de mama.
Las sobrevivientes tenían una probabilidad 5 veces mayor de morir por otras causas relacionadas con la salud, incluso otras NMS y enfermedad cardiovascular o pulmonar (CRI, 5,5; IC 95 %, 3,4–9,0).
La incidencia acumulada de un segundo cáncer de mama asincrónico se elevó de manera significativa, en comparación con los controles (a los 5 años, un 8,0 % entre los sobrevivientes de cáncer infantil vs. un 2,7 % entre los controles; P < 0,001).
Aunque la evidencia disponible en la actualidad es insuficiente para demostrar un beneficio en la supervivencia desde que se inició la vigilancia de cáncer de mama en las mujeres tratadas con radioterapia torácica por un cáncer infantil, es posible que las intervenciones para promover la detección de tumores pequeños y en estadio temprano mejoren el pronóstico; en particular, para quienes tengan opciones de tratamiento más limitadas debido a una exposición previa a la radiación o a las antraciclinas.
En apoyo de la vigilancia, los investigadores del SJLIFE observaron que, en comparación con los cánceres de mama detectados por hallazgos físicos, fue más probable que los cánceres de mama detectados mediante imágenes o mastectomía profiláctica fueran carcinomas in situ, de masas más pequeñas (3,3 cm de tamaño tumoral medio detectado por examen físico vs. 1,1 cm detectado por imágenes), sin compromiso de los ganglios linfáticos y que se trataran sin quimioterapia.[25]
Los investigadores utilizaron datos del CCSS y 2 modelos de simulación de cáncer de mama de la Red de Modelos de Intervención y Vigilancia del Cáncer para estimar los beneficios clínicos, los daños y la rentabilidad de los exámenes de detección del cáncer de mama entre los sobrevivientes de cáncer infantil que habían sido tratados con radiación dirigida al tórax.[55]
Se anticipa que la detección con mamografía e imágenes por resonancia magnética (IRM), como se recomienda en las directrices del COG Long-Term Follow-Up, evitará la mitad de las muertes por cáncer de mama esperadas entre las sobrevivientes de riesgo alto.
En un programa anual, un sobreviviente obtendrá un promedio de 4 o 5 resultados positivos falsos en los exámenes de detección, y 1 a 2 resultados benignos de biopsias a lo largo de su vida.
Debido a los grandes beneficios de supervivencia, se consideró que el equilibrio entre daños y beneficios para los sobrevivientes era apropiado, lo que resultó en una relación entre daños y beneficios más favorable.
En otra investigación del CCSS se cuantificó la relación entre los cambios temporales en el tratamiento del cáncer durante 3 décadas y el riesgo subsiguiente de cáncer de mama.[36]
La incidencia acumulada de cáncer de mama fue del 8,1 % (IC 95 %, 7,3–9,0 %) a los 45 años en 11 550 mujeres sobrevivientes (mediana de edad, 34,2 años), lo que representa un exceso de riesgo 6 veces mayor (CIE, 6,6; IC 95 %, 6,1–7,2), en comparación con controles emparejados por edad, sexo y año calendario.
Al mismo tiempo que los cambios en el tratamiento por década, como la reducción de las tasas de radioterapia torácica y pélvica, el aumento de las tasas de exposición a la quimioterapia con antraciclinas, y el ajuste por edad actual y edad en el momento del diagnóstico, la tasa de cáncer de mama invasivo disminuyó en un 18 % por cada 5 años de edad del diagnóstico primario de cáncer (razón de tasas, 0,82; IC 95 %, 0,74–0,90). Este hallazgo se relacionó en gran medida con la reducción de la tasa de radioterapia dirigida al tórax.
Cáncer de tiroides
El cáncer de tiroides se observa después de las siguientes intervenciones:[12,24,56,57]
Radioterapia dirigida al cuello por linfoma de Hodgkin, leucemia linfoblástica aguda (LLA) y tumores de encéfalo.
Tratamiento con yodo I 131-metayodobenzilguanidina (131I-MIBG) para un neuroblastoma.
ICT para un trasplante de células madre hematopoyéticas.
Quimioterapia sola, sin radiación terapéutica.
La incidencia acumulada a 25 años del cáncer de tiroides entre los sobrevivientes de cáncer infantil es del 0,5 %.[13] Se notificó que el riesgo de cáncer de tiroides entre los sobrevivientes de cáncer infantil es más de 10 veces el de la población general (CIE, 10,5; IC 95 %, 9,1–12).[4] Entre los modificadores importantes del riesgo de cáncer de tiroides relacionado con la radiación se incluyen los siguientes:[28,58]
Sexo femenino.
Menor edad en el momento de la exposición.
Mayor lapso transcurrido desde la exposición.
Dosis de radiación. Se observa una relación lineal entre la dosis y la respuesta de la exposición a la radiación y el cáncer de tiroides hasta una dosis de 10 Gy, luego la relación se estabiliza entre 10 y 30 Gy, y cuando las dosis son más altas se produce una disminución en la OR, en especial en niños menores de 10 años en el momento del tratamiento, lo que indica un efecto citolítico en las células tratadas con dosis más altas.[24,28,59]
En un estudio de casos y controles de los Países Bajos, los sobrevivientes de cáncer infantil con cáncer de tiroides subsiguiente tuvieron más probabilidad que la población general de presentar tumores más pequeños y tumores bilaterales. Los desenlaces del tratamiento fueron similares entre los cánceres de tiroides subsiguientes y esporádicos.[60]
Para obtener información sobre la detección de nódulos tiroideos y cáncer de tiroides, consultar la sección Nódulos tiroideos.
Tumores del sistema nervioso central
Los tumores del sistema nervioso central (SNC) subsiguientes representan un conjunto de subtipos histológicos que abarca desde los gliomas de grado alto hasta los meningiomas benignos. En una revisión exhaustiva del Pediatric Normal Tissue Effects in the Clinic (PENTEC) se analizó el riesgo de NS. En el estudio se notificó una mediana del período de latencia de 10 años para la presentación de una neoplasia maligna del SNC y una mediana del período de latencia de 21 años para la presentación de un meningioma.[61] Las opiniones y prácticas variables con respecto a las neuroimágenes, en comparación con la vigilancia de los síntomas en los sobrevivientes a largo plazo tratados con radiación craneal, no permiten la evaluación exacta de la prevalencia de las lesiones de grado bajo y las lesiones benignas. Por lo tanto, es probable que la prevalencia de estos tumores sea más alta de lo demostrado.
Aparecen tumores de encéfalo después de la irradiación craneal dirigida a otro tipo histológico de tumor de encéfalo o cuando se usa para el control de la enfermedad en pacientes de LLA o linfoma no Hodgkin.[62] Los CIE notificados para las neoplasias subsiguientes del SNC después del tratamiento del cáncer infantil oscilan entre 8,1 y 52,3 en todos los estudios.[24]
El riesgo de tumores de encéfalo subsiguientes también muestra una relación lineal con la dosis de radiación.[26,61,63]
El riesgo de meningioma después de la radiación aumenta con la dosis de radiación[9,61] y, en algunos estudios, se potencia aún más con el aumento de la exposición a metotrexato intratecal;[26] sin embargo, esta conclusión no se ha repetido en forma uniforme.[10]
También se notificaron cavernomas con notable frecuencia después de la irradiación dirigida al SNC, pero se especuló que son consecuencia de procesos angiogénicos en lugar de una verdadera carcinogénesis.[64-66]
Los investigadores del Dutch Long-Term Effects after Childhood Cancer (LATER) describieron las características clínicas de los sobrevivientes de cáncer infantil que presentaron meningiomas confirmados mediante análisis histológico.[11]
De los 6015 sobrevivientes de cáncer infantil de la cohorte LATER, entre los que 1551 se sometieron a radioterapia craneal previa, 93 sobrevivientes de cáncer infantil presentaron meningiomas.
De estos pacientes, 89 (95,7 %) habían recibido radioterapia craneal. La mediana de edad en el momento del diagnóstico fue de 31,8 años (intervalo, 13,2–50,5 años).
De los sobrevivientes, 30 presentaron meningiomas sincrónicos, y 84 tuvieron síntomas. Solo el 16 % de los meningiomas se detectaron en consultorios o clínicas de efectos tardíos.
Todos los sobrevivientes se sometieron a cirugía, y un tercio de ellos (n = 31) recibió también radioterapia. De los sobrevivientes, 12 se sometieron por lo menos a 3 cirugías por crecimiento del tumor residual, recidivas y nuevos meningiomas. Aunque no se describió el grado de resección quirúrgica, las indicaciones de radioterapia durante el seguimiento tras la resección quirúrgica incluyeron tumor residual, recidivas, localización o nuevo meningioma. Durante el seguimiento, 38 sobrevivientes (40,9 %) presentaron nuevos meningiomas, 22 (23,7 %) tuvieron recidivas y al menos 4 murieron a causa del meningioma.
Los investigadores de los European PanCare Childhood and Adolescent Cancer Survivor Care and Follow-Up Studies (PanCareSurFup) describieron los hallazgos en los sobrevivientes de cáncer infantil que presentaron meningiomas o gliomas:[67]
De 69 460 sobrevivientes, 279 presentaron gliomas y 761 presentaron meningiomas.
Se observaron gliomas y meningiomas con mayor frecuencia después de cualquier tumor del SNC o leucemia, lo que representó más del 70 % de todos los gliomas observados y el 80 % de todos los meningiomas.
De los pacientes con estado de radiación craneal conocido que presentaron un glioma, el 61 % había recibido radioterapia craneal previa. Entre los pacientes que presentaron un meningioma, el 64,7 % había recibido radioterapia craneal.
Los sobrevivientes de tumores del SNC que recibieron tratamiento con radioterapia craneal presentaron un mayor riesgo de presentar gliomas, con un riesgo 27 veces superior al de la población general (IC 95 %, 21,5–33,2). Los sobrevivientes de tumores del SNC que no recibieron radioterapia craneal tuvieron un riesgo 8,3 veces superior al de la población general (IC 95 %, 4,9–14,1).
Entre los sobrevivientes de tumores del SNC, el riesgo relativo de los pacientes que recibieron radioterapia craneal fue 13 veces superior al de los pacientes que no recibieron radioterapia craneal (IC 95 %, 6,7–25,4). Entre los sobrevivientes de leucemia, los que recibieron radioterapia craneal tenían un riesgo 5 veces mayor, en comparación con los tratados sin radioterapia craneal (IC 95 %, 2,9–9,9).
En un análisis del PENTEC de NS del SNC se incluyeron 32 estudios publicados de 1035 meningiomas posteriores después de radioterapia previa en sobrevivientes de cáncer infantil.[61]
Se notificó una relación dosis-respuesta significativa para los meningiomas subsiguientes del SNC (exceso de riesgo relativo [ERR]/Gy, 0,44).
La menor edad en el momento del diagnóstico primario se relacionó con un mayor riesgo de meningiomas posteriores.
Las mujeres presentaron un mayor riesgo de meningiomas posteriores que los hombres (oportunidad relativa, 1,46; P < 0,0001).
La mediana del tiempo de latencia hasta la presentación de meningiomas fue de 21 años.
Es posible que las secuelas neurológicas asociadas con los meningiomas incluyan convulsiones, deficiencias auditivas, vestibulares y visuales, disfunción neurológica focal y cefaleas graves.[9] Pese a que se conoce el aumento del riesgo de neoplasias subsiguientes en el SNC en los sobrevivientes de cáncer infantil tratados con irradiación craneal, y ha aumentado el reconocimiento de la morbilidad asociada, la bibliografía médica actual es insuficiente para evaluar los posibles daños y beneficios del uso rutinario de exámenes de detección para estas lesiones.[68] La decisión de iniciar la vigilancia se debe tomar de manera conjunta entre el sobreviviente de cáncer y el proveedor de atención de la salud, después de un análisis profundo en el que se consideren los riesgos y beneficios de la vigilancia de las neoplasias del SNC, como un meningioma.
La radioterapia de protones para el meduloblastoma infantil se relaciona con tasas bajas de lesión del tronco encefálico y neoplasias malignas secundarias. Se notificaron efectos a largo plazo en 178 pacientes pediátricos con meduloblastoma que recibieron tratamiento con cirugía, radioterapia de protones y quimioterapia entre 2002 y 2016 (mediana de seguimiento, 9,3 años).[69]
A los 10 años, 8 pacientes (4,5 %) presentaron un tumor secundario (benigno o maligno) al cabo de una mediana de seguimiento de 9,1 años. De los pacientes, 3 (1,7 %) presentaron una neoplasia maligna secundaria.
Las segundas neoplasias malignas en el campo fueron glioblastoma (n = 2), meningioma (n = 2) y glioma de grado alto (n = 1). Las otras neoplasias malignas fueron una de cada caso: fibroma ovárico, fibromixoma plexiforme de esófago y carcinoma de tiroides papilar.
De los pacientes 4 presentaron lesión del tronco encefálico en una mediana de 4,2 años. La incidencia acumulada a 5 años de lesión del tronco encefálico fue del 1,1 % y la incidencia acumulada a 10 años fue del 1,9 %.
Tumores de hueso y de tejido blando
Los sobrevivientes de retinoblastoma hereditario, sarcoma de Ewing y otros tumores de hueso malignos tienen un riesgo particularmente alto de presentar tumores de hueso y de tejido blando subsiguientes.[70-74]
La radioterapia presenta una relación lineal entre la dosis y la respuesta.[75,76]
En una investigación del PENTEC sobre neoplasias malignas subsiguientes, el ERR/Gy conjunto estimado para sarcomas subsiguientes fue de 0,045. Hubo una posible respuesta a la dosis bifásica, con mayores tasas de recaída y sarcomas posteriores por encima de 55 Gy. Después de administrar 20 Gy y antraciclinas, se prevé que el exceso de riesgo absoluto sea del 0,24 % a los 50 años y del 0,86 % a los 75 años. La mediana del tiempo de latencia hasta la presentación de sarcomas fue de 11 años (intervalo, 4–23 años).[61]
Después del ajuste por radioterapia, los tratamientos con alquilantes [13,61] y antraciclinas [77] se relacionaron con el sarcoma; este riesgo aumenta con la exposición acumulada a estos fármacos.[77]
Hay varios subtipos histológicos de sarcomas de tejido blando, como los sarcomas de tejido blando no rabdomiosarcomatosos, el rabdomiosarcoma, los tumores malignos de vaina del nervio periférico, los tumores de Ewing o neuroectodérmicos primitivos, así como otros tipos de tumores raros.
Evidencia (exceso de riesgo de tumores de hueso y de tejido blando):
En un estudio de casos y controles anidado se incluyeron 228 casos y 228 controles emparejados dentro de una cohorte de 69 460 sobrevivientes a 5 años de cáncer. En el estudio se investigó el riesgo de cáncer de hueso primario subsiguiente de acuerdo a los diferentes grados de exposición a la radiación acumulada, y la relación dosis-respuesta según los diferentes tipos específicos de quimioterapia.[78]
En comparación con el tejido óseo que no se expuso, la OR relacionada con el tejido óseo que se expuso a entre 1 y 4 Gy fue 4,8 veces superior (IC 95 %, 1,2–19,6). La OR relacionada con el tejido óseo que se expuso a entre 5 y 9 Gy fue 9,6 veces superior (IC 95 %, 2,4–37,4). La OR aumentó de manera linear con el incremento de las dosis de radiación (Pde tendencia < 0,001), hasta 78 veces (IC 95 %, 9,2–669,9) para dosis de 40 Gy o más.
Para las dosis de alquilantes acumuladas de 10 000 a 19 999 mg/m2 y 20 000 mg/m2 o más, las OR ajustadas por radiación fueron de 7,1 (IC 95 %, 2,2–22,8) y 8,3 (IC 95 %, 2,8–24,4), respectivamente. Hubo contribuciones independientes de la exposición a procarbazina, ifosfamida o ciclofosfamida.
En un estudio poblacional de 69 460 sobrevivientes a 5 años de cáncer diagnosticado antes de los 20 años de edad, se observaron los siguientes resultados:[71,72]
El riesgo de un cáncer de hueso primario subsiguiente fue 22 veces mayor que el de la población general; con una incidencia acumulada a 45 años del 0,6 %, en comparación con la tasa esperada del 0,03 % en la población general.[71]
El exceso de casos de cáncer de hueso primario subsiguiente disminuyó con la edad y los años transcurridos desde el diagnóstico.[71]
El riesgo de un sarcoma de tejido blando subsiguiente fue casi 16 veces mayor que el de la población general, con una incidencia acumulada a 45 años del 1,4 %, en comparación con una tasa esperada del 0,1 % en la población general.[72]
La mediana de tiempo desde el diagnóstico hasta la presentación de un sarcoma de tejido blando fue de 19 años.[72]
Los sarcomas de tejido blando más frecuentes fueron leiomiosarcomas, neoplasias fibromatosas y tumores malignos de vaina del nervio periférico.[72]
El CIE de los sarcomas primarios fibromatosos subsiguientes disminuyó con el aumento de los años transcurridos desde el diagnóstico y la edad alcanzada, mientras que el CIE del leiomiosarcoma y los tumores malignos de vaina de nervio periférico permanecieron altos en forma uniforme durante todos los años transcurridos desde el diagnóstico y en todas las edades.[72]
Por lo general, el exceso de riesgo absoluto para todos los subtipos de sarcoma fue bajo, excepto para el leiomiosarcoma posterior a un diagnóstico de retinoblastoma (exceso de riesgo absoluto, 52,7 por 10 000 años-persona entre los sobrevivientes de 45 años o más desde el diagnóstico).[72]
El riesgo de presentar un leiomiosarcoma fue 30 veces más alto en los sobrevivientes de cáncer infantil, en comparación con un exceso de riesgo de 0,7 para la población general.[72]
Los sobrevivientes de retinoblastoma tuvieron el riesgo más alto (CIE, 342,9), seguidos por los sobrevivientes de tumor de Wilms (CIE, 74,2).
De los leiomiosarcomas observados después de un diagnóstico de tumor de Wilms, el 90 % se presentaron dentro del tejido irradiado.
En una cohorte del CCSS, un aumento del riesgo de sarcoma óseo o de tejido blando subsiguientes se relacionó con la radioterapia, un diagnóstico primario de sarcoma, antecedentes de otras NS y un tratamiento con dosis más altas de antraciclinas o alquilantes.[27]
La incidencia acumulada a 30 años de un sarcoma subsiguiente en los participantes del CCSS fue del 1,08 % para los sobrevivientes que recibieron radioterapia y del 0,5 % para los sobrevivientes que no recibieron radioterapia.
Se usó un modelo de riesgo según la dosis para estudiar el riesgo de sarcoma óseo en una cohorte retrospectiva de 4171 sobrevivientes de cáncer infantil sólido tratados entre 1942 y 1986 (mediana de seguimiento, 26 años).[75]
Los resultados demostraron que el riesgo de sarcoma óseo aumentó de manera leve hasta una dosis acumulada de radiación absorbida por los órganos de 15 Gy (CRI, 8,2; IC 95 %, 1,6–42,9) y, luego, se presentó un incremento rápido con las dosis de radiación más altas (CRI de 30 Gy o más, 117,9; IC 95 %, 36,5–380,6), en comparación con los pacientes que no se trataron con radioterapia.
El exceso de riesgo relativo por cada Gy en este modelo fue de 1,77 (IC 95 %, 0,62–5,94).
En los sobrevivientes de retinoblastoma bilateral, el sarcoma es la NS maligna que se observa con más frecuencia, en particular, el osteosarcoma.[79-81] La contribución de la quimioterapia a la carcinogénesis de tumores malignos sólidos se destacó en un estudio de seguimiento a largo plazo de 906 sobrevivientes de retinoblastoma hereditario a 5 años diagnosticados entre 1914 y 1996 y observados hasta 2009.[70]
El tratamiento con alquilantes aumentó de forma significativa el riesgo de tumores óseos subsiguientes (CRI, 1,60; IC 95 %, 1,03–2,49) y leiomiosarcoma (CRI, 2,67; IC 95 %, 1,22–5,85) entre los miembros de la cohorte.
Los casos de leiomiosarcoma fueron más comunes luego de la quimioterapia con alquilantes y radioterapia, en comparación con la radioterapia sola (5,8 vs.1,6 % a los 40 años de edad; P = 0,01).
En el CCSS se notificó el seguimiento de 105 casos y 422 controles emparejados en un estudio de casos y controles anidado de una cohorte de 14 372 sobrevivientes de cáncer infantil:[77]
Los sarcomas de tejido blando se presentaron en una mediana de 11,8 años (intervalo, 5,3–31,3 años) desde el diagnóstico original.
Cualquier exposición a la radiación se relacionó con un aumento del riesgo de sarcoma de tejido blando (OR, 4,1; IC 95 %, 1,8–9,5), que mostró una relación lineal entre la dosis y la respuesta.
La exposición a las antraciclinas se relacionó con riesgo de sarcoma de tejido blando (OR, 3,5; IC 95 %, 1,6–7,7), con independencia de la dosis de radiación.
En una cohorte de 952 sobrevivientes de retinoblastoma hereditario diagnosticado entre 1914 y 2006 que recibieron irradiación, los investigadores del CCSS observaron que el riesgo elevado de sarcomas óseos y de tejido blando variaba según la edad, la ubicación y el sexo.[81]
Los sarcomas óseos de cabeza y cuello y los sarcomas de tejido blando se diagnosticaron desde la primera infancia y continuaron hasta la edad adulta (incidencia acumulada a 60 años del 6,8 % y el 9,3 %, respectivamente).
La incidencia de los sarcomas óseos de cuerpo y extremidades se redujo después de la adolescencia (incidencia acumulada a 60 años, 3,5 %).
La incidencia de sarcoma de tejido blando de cuerpo y extremidades fue muy baja hasta los 30 años, momento en que la incidencia aumentó de manera brusca (incidencia acumulada a 60 años, 6,6 %), en especial, en las mujeres (incidencia acumulada a 60 años, 9,4 %).
En un estudio retrospectivo de 160 pacientes con retinoblastoma hereditario que recibieron radioterapia no se determinó ninguna correlación entre la edad (antes o después de los 12 meses) a la que se administró la radioterapia de haz externo y la formación de una neoplasia maligna subsiguiente.[73]
Los pacientes con una neoplasia maligna subsiguiente o sin esta, no diferían en cuanto al tipo de variante de RB1. Además, no hubo relación entre el tipo de variante y la ubicación de la NMS, o el tipo de neoplasia y la edad en el momento del diagnóstico.
En el estudio se demostró que los pacientes con una variante de penetrancia baja que reciben radioterapia de haz externo continúan presentando riesgo de NMS y se les debe realizar un seguimiento minucioso.
Cáncer de piel
Los carcinomas queratinocíticos (por ejemplo, carcinomas de células basales [CCB] y carcinomas de células escamosas [CCE], también llamados cánceres de piel no melanoma [CPNM]) son algunas de las NS más comunes entre los sobrevivientes de cáncer infantil y exhiben una fuerte relación con la radioterapia.[4,82,83] La observancia de conductas de protección solar reduce la exposición a la radiación UV que exacerba el riesgo.
El CCSS llevó a cabo un ensayo aleatorizado, controlado y de eficacia comparativa con el fin de evaluar métodos para mejorar la detección temprana del cáncer de piel en los sobrevivientes de cáncer infantil de riesgo alto después de la exposición a la radioterapia. Los participantes se asignaron al azar al grupo experimental, que recibió materiales impresos en combinación con estrategias mHealth (mensajes de texto y uso del sitio web Advancing Survivors’ Knowledge), o al grupo de control. La tasa de exámenes de detección mejoró 1,5 veces en el grupo experimental. En los 3 grupos de intervención las tasas de examen médico de la piel aumentaron desde el inicio hasta los 12 meses, y las tasas de autoexamen aumentaron desde el inicio hasta los 18 meses. Sin embargo, el aumento de las tasas no fue diferente entre los grupos de intervención.[84]
Evidencia (exceso de riesgo de carcinomas queratinocíticos:
En un análisis actualizado del CCSS se confirmó un aumento del riesgo de carcinomas queratinocíticos en aquellos participantes que en el pasado habían recibido radioterapia. En comparación con los participantes que no recibieron radioterapia, los participantes del CCSS tratados con radioterapia presentaron un riesgo 4,5 veces mayor de carcinomas queratinocíticos (IC 95 %, 3,5–5,9)[83].
El riesgo de carcinomas queratinocíticos aumentó a medida que aumentaron las dosis de radioterapia. Cada 10 Gy de radioterapia en el sitio del carcinoma queratinocítico, el RR fue similar para los sitios en cabeza y cuello (1,0; IC 95 %, 1,0–1,1) y extremidades (1,0; IC 95 %, 0,7–1,3). El RR fue levemente superior en los sitios del tronco (1,1; IC 95 %, 1,0–1,3).
Los TCMH alogénicos y autógenos se relacionaron con un aumento del riesgo de carcinomas queratinocíticos (RR para el TCMH alogénico, 3,4; IC 95 %, 2,4–4,8 y RR para el TCMH autógeno, 2,3; IC 95 %, 1,2–4,2).
En la cohorte de 5843 sobrevivientes de cáncer infantil del DCOG-LATER, los investigadores encontraron que los sobrevivientes de cáncer infantil tenían un riesgo 30 veces mayor de presentar CCB.[30]
Después del primer diagnóstico de CCB, el 46,7 % de los pacientes presentaron otros CCB.
El riesgo de CCB se relacionó con cualquier radioterapia dirigida al campo de radiación pertinente (sitio de CCB) (CRI, 14,32) y con el porcentaje calculado del área de superficie de la piel expuesta (26–75 %). CRI, 1,99; 76–100 %: CRI, 2,16 vs. 1–25 % expuesta; Pde tendencia entre los expuestos = 0,002).
El riesgo de CCB no se relacionó con la dosis prescrita de radiación ni la probabilidad del área de piel de exponerse al sol.
De todos los grupos de quimioterapia examinados, solo los alcaloides de vinca aumentaron el riesgo de CCB (CRI, 1,54).
Se ha notificado que la presentación de un carcinoma queratinocítico como primera NS identifica a una población en riesgo alto de NS maligna invasiva en el futuro.[7]
Los investigadores del CCSS observaron una incidencia acumulada de neoplasias malignas del 20,3 % (IC 95 %, 13,0–27,6 %) a los 15 años en sobrevivientes expuestos a radiación que presentaron un carcinoma queratinocítico como primera NS, en comparación con el 10,7 % (IC 95 %, 7,2–14,2 %) en aquellos cuya primera NS fue una neoplasia maligna invasiva.
También se notificó melanoma maligno como una NS en cohortes de niños sobrevivientes de cáncer infantil, si bien con una incidencia mucho más baja que los CPNM.
Los factores de riesgo del melanoma maligno identificados en estos estudios son los siguientes:[85]
Radioterapia (>40 Gy).
Alquilantes.
Bleomicina.
Evidencia (exceso de riesgo de melanoma):
Un grupo de investigadores evaluaron la incidencia, los factores de riesgo y los desenlaces de pacientes con melanoma en la cohorte del CCSS (n = 25 716).[85]
Hubo 160 sobrevivientes (mediana de edad alcanzada, 33 años) que presentaron 177 melanomas (110 invasivos, 62 cutáneos in situ, 5 oculares).
La incidencia acumulada a 40 años fue del 1,1 % en todos los participantes y del 1,5 % en aquellos que recibieron dosis acumuladas de radiación de 40 Gy o superiores. El cociente de incidencia estándar fue de 2,0 cuando se lo comparó con el de la población general.
Un aumento de melanomas cutáneos se relacionó con dosis acumuladas de radiación de 40 Gy o más en el sitio del melanoma (CRI, 2,0), dosis equivalentes de ciclofosfamida acumuladas de 20 000 mg/m2 o más (CRI, 1,9) y exposición a la bleomicina (CRI, 2,2).
La presencia de un melanoma invasivo se relacionó con el doble de riesgo de muerte.
En una revisión sistemática, que incluyó datos de 19 estudios originales (n total = 151 575 sobrevivientes; mediana de seguimiento de 13 años), se observó una incidencia anual de 10,8 casos de melanoma maligno por 100 000 sobrevivientes de cáncer infantil.[86]
Los melanomas se presentan con mayor frecuencia en los sobrevivientes de linfoma de Hodgkin, retinoblastoma hereditario, sarcoma de tejido blando y tumores gonadales; sin embargo, el número relativamente pequeño de sobrevivientes representados en los estudios importantes impide la evaluación del riesgo de melanoma entre otros tipos de cáncer infantil.
Riesgo de cáncer de piel después de retinoblastoma
La incidencia de melanoma y CPNM se evaluó en una cohorte de 1851 sobrevivientes blancos a largo plazo de retinoblastoma (1020, hereditario y 831, no hereditario) diagnosticado entre 1914 y 2006, que se vigilaron hasta 2016.[87]
De todos los pacientes, 33 sobrevivientes de retinoblastoma hereditario y 7 sobrevivientes de retinoblastoma no hereditario presentaron melanoma, mientras que 26 sobrevivientes de retinoblastoma hereditario y 9 sobrevivientes de retinoblastoma no hereditario, presentaron CPNM. La mediana de edad de la aparición del cáncer de piel fue alrededor de 20 años más temprana en los sobrevivientes de retinoblastoma hereditario, en comparación con los sobrevivientes de retinoblastoma no hereditario.
La mayoría de los CPNM se presentaron en la cabeza y el cuello, mientras que los melanomas estaban repartidos en una zona más amplia, de manera similar a la distribución que se observa en las familias propensas al melanoma.
Al cabo de 50 años desde el diagnóstico de retinoblastoma, en los sobrevivientes de retinoblastoma hereditario la incidencia acumulada de melanoma fue del 4,5 % y la incidencia acumulada de CPNM fue del 3,7 %. En los sobrevivientes de retinoblastoma no hereditario, la incidencia acumulada de melanoma fue del 0,7 % y la incidencia acumulada de CPNM fue del 1,5 %.
Cáncer de pulmón
Entre las cohortes de sobrevivientes de cáncer infantil, el cáncer de pulmón representa una NS relativamente poco frecuente. En una revisión sistemática del PENTEC se notificó una mediana del período de latencia de 25 años (intervalo, 19–29 años) desde el diagnóstico de cáncer infantil hasta la aparición de una NS pulmonar.[61] El ERR/Gy fue de 0,068, en comparación con los riesgos acumulados subyacentes. Después de la administración de 20 Gy, se predijo un exceso de riesgo absoluto del 0,27 % a los 50 años. Después de 50 Gy, se predijo un exceso de riesgo absoluto del 0,7 % a los 50 años.[61]
Evidencia (exceso de riesgo de cáncer de pulmón):
La incidencia acumulada a 30 años de cáncer de pulmón entre los participantes del CCSS fue del 0,16 % (IC 95 %, 0,09–0,23 %), lo que representa un CIE de 4,0 (IC 95 %, 2,9–5,4).[88]
El riesgo de cáncer de pulmón demostró una relación con la dosis de radiación torácica: 10 a 30 Gy (CRI, 3,4; IC 95 %, 9,1–11,0), más de 30 a 40 Gy (CRI, 4,6; IC 95 %, 1,5–14,3) y más de 40 Gy (CRI, 1,05; IC 95 %, 10–27,0).
La incidencia acumulada de NMS pulmonares fue más alta en los fumadores o exfumadores, en comparación con aquellas personas que nunca habían fumado.
El CIE para el cáncer de pulmón fue más alto entre los sobrevivientes de linfoma de Hodgkin (CIE, 9,3; IC 95 %, 6,2–13,4) y cáncer de hueso (CIE, 4,4; IC 95 %, 1,8–9,1).
La incidencia acumulada a 25 años de cáncer de pulmón en la cohorte del DCOG-LATER fue del 0,1 % (IC 95 %, 0,0–0,3 %).[13]
La incidencia fue alrededor de 4 veces mayor de lo que se esperaría en la población general (CIE, 4,3; IC 95 %, 1,9–8,5).
Se notificó cáncer de pulmón luego de irradiación dirigida al tórax debido a un linfoma de Hodgkin.[89]
El riesgo aumenta conforme lo hace el lapso de tiempo a partir del diagnóstico.
El consumo de tabaco se relacionó con el cáncer de pulmón que se presenta luego de la radioterapia para un linfoma de Hodgkin.[89]
El aumento del riesgo de cáncer de pulmón según se incrementa la dosis de radiación es mayor en los pacientes que fuman luego de exponerse a la radiación que entre aquellos que se abstienen de fumar (P = 0,04).
Cáncer digestivo
Hay evidencia reciente de que los sobrevivientes de cáncer infantil presentan neoplasias malignas digestivas con mayor frecuencia y a una edad más temprana que la población general. Esta evidencia respalda la necesidad de iniciar temprano la vigilancia del carcinoma colorrectal.[90-92]
Evidencia (exceso de riesgo de cáncer digestivo):
En un estudio de casos y controles anidados en una cohorte de Francia y Reino Unido de sobrevivientes de tumores sólidos infantiles diagnosticados antes de los 17 años de edad, el riesgo de presentar una NS de órgano digestivo varió con el tratamiento.[93]
El riesgo de cáncer digestivo fue 9,7 más alto que en los controles de la población.
Las NS comprometieron con más frecuencia el colon o el recto (42 %), el hígado (24 %) y el estómago (19 %).
Se observó una relación estrecha entre la dosis y la respuesta a la radiación, con una OR de 5,2 (IC 95 %, 1,7–16,0) para una dosis de radiación local de 10 a 29 Gy, y de 9,6 (IC 95 %, 2,6–35,2) para una dosis de 30 Gy o superior, en comparación con los sobrevivientes que no habían recibido radioterapia.
La quimioterapia sola y las modalidades de tratamiento combinado se relacionaron con un aumento significativo del riesgo de presentar una NS digestiva (CIE, 9,1; IC 95 %, 2,3–23,6; CIE, 29,0; IC 95 %, 20,5–39,8, respectivamente).
El consorcio de los estudios PanCare Childhood and Adolescent Cancer Survivor Care y PanCareSurFup (estudio de seguimiento) cuantificó el riesgo absoluto según las características del tratamiento de radioterapia en una cohorte de 69 450 sobrevivientes a 5 años de cáncer infantil en Europa. Las OR se calcularon a partir de un estudio de casos y controles que incluyó 143 cánceres colorrectales subsiguientes dentro de la cohorte de sobrevivientes de cáncer infantil.[92]
Los sobrevivientes que recibieron cualquier radioterapia dirigida al abdomen o la pelvis tuvieron una probabilidad 3 veces superior de presentar un cáncer colorrectal subsiguiente que aquellos que no recibieron radioterapia (OR, 3,1; IC 95 %, 1,4–6,6).
Hacia los 40 años de edad, los sobrevivientes que habían recibido radioterapia dirigida al abdomen o la pelvis tenían un riesgo similar de cáncer colorrectal que las personas de 50 años de la población general, que es cuando comienzan a realizarse los exámenes de detección en la población.
El riesgo de cáncer colorrectal aumenta con la dosis de radioterapia dirigida al abdomen o la pelvis. Los sobrevivientes que se trataron con radioterapia dirigida a la columna vertebral o al abdomen completo tienen un riesgo mayor que aquellos que se trataron con radioterapia dirigida a otros campos del abdomen o la pelvis.
Los investigadores del CCSS notificaron un riesgo 4,6 veces más alto de NS digestiva entre los participantes del estudio que en la población general (IC 95 %, 3,4–6,1).[90]
Las NS comprometieron con más frecuencia el colon (39 %), el recto o el ano (16 %), el hígado (18 %) y el estómago (13 %).
El CIE para cáncer colorrectal fue de 4,2 (IC, 2,8–6,3).
El tipo histológico de NS gastrointestinal más prevalente fue el adenocarcinoma (56 %).
El riesgo más alto de las NS digestivas se relacionó con la irradiación abdominal (CIE, 11,2; IC, 7,6–16,4), pero los sobrevivientes no expuestos a la radiación también presentaron un aumento significativo del riesgo (CIE, 2,4; IC, 1,4–3,9).
Las dosis altas de procarbazina (riesgo relativo [RR], 3,2; IC 1,1–9,4) y derivados del platino (RR, 7,6; IC, 2,3–25,5) aumentaron de manera independiente el riesgo de NS digestiva.
Los investigadores del St. Jude Children's Research Hospital observaron que el CIE para el carcinoma colorrectal subsiguiente fue de 10,9 (IC 95 %, 6,6–17,0), en comparación con los controles de la población de los Estados Unidos. Los investigadores también observaron lo siguiente:[91]
La incidencia de un carcinoma colorrectal subsiguiente aumentó de forma pronunciada conforme el avance de la edad, con una incidencia acumulada a los 40 años de 1,4 ± 0,53 % en la cohorte completa (n = 13 048) y 2,3 ± 0,83 % en los sobrevivientes a 5 años.
El riesgo de carcinoma colorrectal aumentó en un 70 % con cada aumento de 10 Gy en la dosis de radiación. El incremento del volumen de radiación también aumentó el riesgo.
El tratamiento de quimioterapia con alquilantes también se vinculó con un exceso de riesgo de 8,8 veces de carcinoma colorrectal subsiguiente.
En un estudio prospectivo multinstitucional, se observó que se encontraron pólipos neoplásicos precancerosos en el 27,8 % de los sobrevivientes de cáncer infantil que recibieron radiación dirigida al abdomen o la pelvis por lo menos 10 años antes y que se sometieron a una colonoscopia de detección entre los 35 y los 49 años.[94]
La prevalencia de pólipos es, por lo menos, tan alta como la notificada con anterioridad para la población de riesgo medio mayor de 50 años y es similar a la tasa de incidencia de un 24 % para los pacientes de cáncer de colon no polipósico hereditario. No se conocen las tasas de prevalencia de pólipos en la población general de personas entre los 35 a 49 años.
En un estudio de vinculación de registros del DCOG-LATER se evaluó el riesgo de adenomas colorrectales confirmados por evaluación histológica en 5843 sobrevivientes a 5 años de cáncer infantil seguidos durante una mediana de 24,9 años.[95]
La incidencia acumulada de adenoma colorrectal a los 45 años fue de un 3,6 % en los sobrevivientes que recibieron radiación abdominal pélvica versus un 2,0 % en los sobrevivientes que no recibieron este tipo de radiación, versus un 1,0 % en los hermanos.
Los factores relacionados con riesgo de adenoma fueron la radiación abdominal pélvica (CRI, 2,1), la ICT (CRI, 10,6), el uso de cisplatino (CRI, 2,1 para <480 mg/m2; CRI, 3,8 para ≥480 mg/m2), el diagnóstico de hepatoblastoma (CRI, 27,1) y los antecedentes familiares de cáncer colorrectal de inicio temprano (CRI, 20,5).
La exposición a procarbazina también se relacionó con aumento del riesgo en sobrevivientes no expuestos a radiación abdominal pélvica ni ICT (CRI, 2,7).
El consorcio PanCareSurFup informó sobre cánceres digestivos en una cohorte de 69 460 sobrevivientes a 5 años de cáncer infantil en Europa.[96]
Los sobrevivientes de tumor de Wilms (CIE, 12,1; IC 95 %, 9,6–15,1) y linfoma de Hodgkin (CIE, 7,3; IC 95 %, 5,9–9,0) tuvieron el riesgo más alto de NMS digestivas.
A los 55 años, el 2,3 % de los sobrevivientes de tumor de Wilms y linfoma de Hodgkin presentaron un cáncer colorrectal secundario, una tasa comparable a la de la población general con 2 o más familiares de primer grado afectados por cáncer digestivo.
En un estudio de casos y controles anidado se examinó la tasa de cáncer colorrectal de acuerdo con la dosis de radioterapia para el intestino grueso y la dosis de procarbazina en los sobrevivientes de linfoma de Hodgkin a 5 años diagnosticados entre los 15 y 50 años de edad (solo el 27 % de los casos tenían entre 15 y 24 años de edad) en 5 centros hospitalarios de los Países Bajos (diagnosticado entre 1964 y 2000; mediana de seguimiento, 26 años).[97]
La mediana de intervalo entre el diagnóstico de linfoma de Hodgkin y el cáncer colorrectal subsiguiente fue de 25,7 años (intervalo, 18,2–31,6 años).
El tratamiento con radioterapia subdiafragmática (RR, 2,4; IC 95 %, 1,4–4,1) y más de 8,4 g/m2 de procarbazina (RR, 2,5; IC 95 %, 1,3–5,0) se relacionó con un aumento de las tasas de cáncer colorrectal en el análisis univariante.
La tasa de cáncer colorrectal aumentó de manera lineal con la dosis media de radioterapia dirigida a todo el intestino grueso y la dosis dirigida al segmento intestinal afectado. La respuesta a la dosis se hizo más pronunciada con dosis más altas de procarbazina y aumentó 1,2 veces (IC 95 %, 1,1–1,3) por cada aumento de 1 g/m2 en procarbazina.
Cánceres de la boca
El consorcio PanCareSurFup informó sobre los riesgos de segundas neoplasias primarias orales (validadas mediante informes patológicos) en una cohorte de 69 460 sobrevivientes a 5 años de cáncer infantil en Europa.[98]
Segundas neoplasias primarias orales (n = 145; 64 glándula salival, 38 lengua, 20 faringe, 2 labio, y otros 21) en 143 sobrevivientes a una mediana de edad de 32 años. Esto representa un exceso de riesgo 5 veces mayor que el esperado en la población general.
Los grupos de diagnóstico de cáncer infantil con mayor riesgo de una segunda neoplasia primaria oral fueron leucemia (CIE, 19,2; IC 95 %, 14,6–25,2), sarcoma óseo (CIE, 6,4; IC 95 %, 3,7–11,0), linfoma de Hodgkin (CIE, 6,2; IC 95 %, 3,9–9,9) y sarcoma de tejido blando (CIE, 5,0; IC 95 %, 3,0–8,5).
Las relaciones específicas observadas con las segundas neoplasias primarias orales incluyeron radioterapia y neoplasias de glándulas salivales (CIE, 33; IC 95 %, 25,3–44,5), quimioterapia (cualquier exposición) y neoplasias de lengua (CIE, 15,9; IC 95 %, 10,6–23,7).
No se disponía de datos para evaluar la contribución de los comportamientos de salud (consumo de tabaco y de bebidas alcohólicas) y el estado de la infección por el virus del papiloma humano (VPH) a la aparición de segundas neoplasias primarias orales.
Cánceres urogenitales
La aparición de cánceres urogenitales primarios subsiguientes en sobrevivientes de cáncer diagnosticados durante la niñez o adolescencia es poco frecuente.
Con los datos del SEER de 43 991 pacientes (edad <20 años) diagnosticados con un primer cáncer primario entre 1975 y 2016, el riesgo de cáncer del aparato urinario fue más alto tanto para mujeres (CIE, 5,18; IC 95 %, 3,65–7,14) como para hombres (CIE, 2,80; IC 95 %, 1,94–3,92), en comparación con la población general.[99]
Las mujeres tuvieron más probabilidad que los hombres de presentar un cáncer subsiguiente del aparato urinario (CIE, 1,86; IC 95 %, 1,13–3,03) y cáncer de riñón (CIE, 1,97; IC 95 %, 1,11–3,53).
Las mujeres con cualquier primer cáncer tuvieron un riesgo más alto que la población general de presentar cánceres de cuerpo uterino (CIE, 2,32; IC 95 %, 1,49–3,45) y vulva (CIE, 4,27; IC 95 %, 1,38–9,95).
Carcinoma renal
De acuerdo con los informes de sobrevivientes de cáncer que se presenta en la edad adulta, se ha observado un aumento del riesgo de carcinoma renal en sobrevivientes de cáncer infantil.[29,100,101] Es posible que la predisposición genética subyacente también desempeñe una función en el riesgo de presentar carcinomas renales porque se observaron casos infrecuentes de carcinoma renal en niños con esclerosis tuberosa.[100] También se notificaron casos de carcinoma renal secundario relacionados con las translocaciones de Xp11.2 y las fusiones del gen TFE3, lo que indica que la quimioterapia citotóxica tal vez contribuya a la carcinogénesis renal.[102-104]
Evidencia (exceso de riesgo de carcinoma renal):
Los investigadores del CCSS notificaron un exceso de riesgo significativo de carcinoma renal subsiguiente en 14 358 sobrevivientes a 5 años en la cohorte (CIE, 8,0; IC 95 %, 5,2–11,7), en comparación con la población general.[100]
El exceso absoluto total de riesgo notificado de 8,4 por 105 años-persona indica que estos casos son relativamente infrecuentes. El riesgo más alto se observó en los siguientes casos:
Sobrevivientes de neuroblastoma (CIE, 85,8; IC 95 %, 38,4–175,2).[100] Se propuso la hipótesis de que la radioterapia predispone a los niños con neuroblastoma de riesgo alto a presentar carcinoma renal.[105]
Personas tratadas con 5 Gy o más de radioterapia dirigida al riñón (RR, 3,8; IC 95 %, 1,6–9,3).[100]
Personas tratadas con quimioterapia con derivados del platino (RR, 3,5; IC 95 %, 1,0–11,2).[100]
Neoplasias malignas relacionadas con el virus del papiloma humano
Evidencia (neoplasias malignas subsiguientes relacionadas con el virus del papiloma humano [VPH]):
En un estudio del CCSS se evaluó la presencia de tipos de cáncer en los que el VPH es un factor de riesgo etiológico establecido. En el estudio participaron 24 363 sobrevivientes de cáncer infantil que presentaban una mediana de 21 años desde el diagnóstico.[106]
La incidencia acumulada a 30 años de un cáncer relacionado con el VPH fue del 0,3 % (IC 95 %, 0,2–0,4 %), lo que refleja un exceso de riesgo 3 veces superior (CIE, 2,86; IC 95 %, 2,05–4,00) respecto al de la población general.
Los hombres y las mujeres sobrevivientes de cáncer tenían un riesgo elevado de presentar NMS relacionadas con el VPH orofaríngeas (CIE en hombres, 4,06; CIE en mujeres, 8,44) y anorrectales (CIE en hombres, 13,56; CIE en mujeres, 9,16). Sin embargo, las mujeres no tuvieron un riesgo mayor de cáncer de cuello uterino o vulva, en comparación con la población general.
Entre los factores de riesgo independientes para los cánceres relacionados con el VPH identificados mediante un modelo multivariante estaban el sexo masculino (vs. sexo femenino: CIE relativo, 1,99; IC 95 %, 1,00–3,94); dosis de radioterapia dirigida a la cabeza, el cuello y la pelvis superiores a 30 Gy (vs. ninguna dosis: CIE relativo, 2,35; IC 95 %, 1,11–4,97); y dosis equivalentes de cisplatino superiores a 400 mg/m2 (vs. ninguna dosis: CIE relativo, 4,51; IC 95 %, 1,78–11,43).
En un estudio poblacional se usaron los datos del registro Surveillance, Epidemiology, and End Results (SEER) para evaluar los factores de riesgo y la tendencia a presentar NMS relacionadas con el VPH de 374 408 pacientes adolescentes y adultos jóvenes (AAJ) (edad en el momento del diagnóstico: 11 % de 15–24 años, 28 % de 25–34 años y 61 % de 35–44 años). Los pacientes se diagnosticaron entre 1976 y 2015.[107]
La incidencia de NMS relacionadas con el VPH se redujo a lo largo del periodo del estudio y la incidencia general de NMS relacionadas con el VPH entre sobrevivientes de cáncer AAJ fue baja; solo afectó a un 0,4 % de los sobrevivientes.
En los sobrevivientes AAJ, el riesgo de cualquier NMS relacionada con el VPH aumentó un 70 % (CIE, 1,70; IC 95 %, 1,61–1,79) y el riesgo de cáncer de orofaringe aumentó un 117 % (CIE, 2,17; IC 95 %, 2,00–2,35), en comparación con la población general.
En general, el riesgo de cáncer de cuello uterino fue bajo entre las sobrevivientes (CIE, 0,85; IC 95 %, 0,76–0,95), pero las sobrevivientes AAJ hispanas tuvieron un aumento significativo del cáncer de cuello uterino (CIE, 1,46; IC 95 %, 1,01–2,06).
Entre los sobrevivientes con cánceres primarios asociados al VPH, haber recibido quimioterapia y radioterapia se vinculó con cualquier NMS relacionada con el VPH. Sin embargo, esos tratamientos no se vincularon con NMS relacionadas con el VPH entre los sobrevivientes cuyos cánceres primarios no se asociaban al VPH.
Desenlaces de supervivencia después de neoplasias subsiguientes
El desenlace después del diagnóstico de una NS es variable porque es posible que el tratamiento de algunos subtipos histológicos se vea comprometido si el tratamiento del cáncer infantil incluyó dosis acumuladas de fármacos que superó el umbral de tolerancia tisular.
Se compararon personas menores de 60 años que presentaban por primera vez una neoplasia maligna primaria (n = 1 332 203) con sobrevivientes de cáncer infantil (n = 1409) que presentaban una segunda neoplasia maligna primaria utilizando los datos del SEER Program.[108]
Los sobrevivientes de cáncer infantil a los que se les diagnosticó una segunda neoplasia maligna primaria presentaron una supervivencia general más precaria que la de sus pares sin antecedentes de cáncer (CRI, 1,86; IC 95 %, 1,72–2,02) después de que en el estudio se hubiese tenido en cuenta el tipo de cáncer, la edad, el sexo, la raza y la década en la que se hizo el diagnóstico.
El antecedente de cáncer infantil se relacionó de manera constante con un riesgo de muerte 2 a 3 veces mayor por las segundas neoplasias malignas primarias de diagnóstico más frecuente, incluso cáncer de mama, cáncer de tiroides, LMA, cáncer de encéfalo, melanoma, cáncer de hueso y sarcoma de tejido blando.
En un estudio de mujeres participantes en el CCSS, que luego recibieron un diagnóstico de cáncer de mama (n = 274) y se emparejaron con un grupo control de mujeres (n = 1095) con cáncer de mama de nueva aparición, se observó que las sobrevivientes de cáncer infantil tenían tasas de mortalidad elevadas (CRI, 2,2; IC 95 %, 1,7–3,0), incluso después de ajustar según el tratamiento del cáncer de mama.[54]
Las sobrevivientes tenían una probabilidad 5 veces mayor de morir como resultado de otras causas relacionadas con la salud, incluso otras NMS y enfermedad cardiovascular o pulmonar (CRI, 5,5; IC 95 %, 3,4–9,0).
La incidencia acumulada de un segundo cáncer de mama asincrónico se elevó de manera significativa, en comparación con los controles (a los 5 años, 8,0 % entre los sobrevivientes de cáncer infantil vs. 2,7 % entre los controles; P < 0,001).
Neoplasias subsiguientes y susceptibilidad genética
En la bibliografía médica se sustenta de forma clara la función de la quimioterapia y la radioterapia en la presentación de NS. Sin embargo, la variabilidad interindividual indica que la variación genética desempeña una función en la susceptibilidad a exposiciones genotóxicas o que un síndrome de susceptibilidad genética confiere un aumento de riesgo de cáncer; por ejemplo, el síndrome de Li-Fraumeni.[109,110] En un estudio poblacional del Swiss Childhood Cancer Survivor Study los síndromes de predisposición al cáncer se relacionaron con un riesgo alto de segundas neoplasias primarias antes de los 21 años y representaron el factor de riesgo más importante (CRI, 7,8; IC 95 %, 4,8–12,7) de presentar un segundo cáncer primario.[111]
En estudios previos se demostró que los sobrevivientes de cáncer infantil con antecedentes familiares del síndrome de Li-Fraumeni en particular o con antecedentes familiares de cáncer tienen un aumento de riesgo de presentar una NS.[112,113] En un registro prospectivo se hizo un seguimiento a 480 personas con variantes de la línea germinal patogénicas o posiblemente patogénicas en TP53.[114] Se realizó un seguimiento a las personas que presentaron un primer cáncer para detectar si se presentaba una segunda neoplasia maligna. Entre las personas que tenían menos de 17 años en el momento del diagnóstico del primer cáncer, el 50 % presentó un segundo cáncer en el transcurso de 20 años.
Es posible que el riesgo de NS se modifique debido a variantes de los genes de penetrancia alta que conducen a estas enfermedades genéticas graves (por ejemplo, el síndrome de Li-Fraumeni).[113] Sin embargo, se espera que el riesgo atribuible sea muy reducido debido a la prevalencia extremadamente baja de variantes de los genes de penetrancia alta.
De igual manera, los niños con neurofibromatosis de tipo 1 (NF1) que presentan un tumor primario tienen un riesgo mayor de NS, en comparación con los sobrevivientes de cáncer infantil sin NF1. El tratamiento con radiación pero sin alquilantes aumenta el riesgo de NS en los sobrevivientes de NF1.[115] Las NS representan un factor importante en el exceso de mortalidad en adultos sobrevivientes de glioma infantil con NF1.[116] Estos sobrevivientes presentaron NMS de inicio tardío (>5 años desde el diagnóstico) a una tasa 4 veces superior a la de los sobrevivientes de glioma sin NF1 (4,02; intervalo, 2,12–7,62). La tasa de mortalidad tardía por todas las causas a 30 años fue del 46,3 % (IC 95 %, 23,9–62,2 %) en los sobrevivientes de glioma con NF1, en comparación con el 18 % (IC 95 %, 16,1–20,0 %) en los sobrevivientes de glioma sin NF1. Las causas más comunes de muerte entre los sobrevivientes con NF1 y glioma fueron las NS.
En el Cuadro 1 se resume el conjunto de neoplasias, los genes afectados y el modelo mendeliano de herencia de síndromes específicos de predisposición hereditaria al cáncer.
Cuadro 1. Síndromes específicos de predisposición hereditaria al cáncera
Síndrome
Tipos principales de tumor
Gen afectado
Moo de herencia
LMA = leucemia mieloide aguda; SMD = síndromes mielodisplásicos; WAGR = tumor de Wilms, aniridia, anormalidades genitourinarias y una variedad de alteraciones en el desarrollo neurológico.
bDominante en una fracción de pacientes; se pueden presentar variantes espontáneas.
Poliposis adenomatosa del colon
Cáncer de colon, hepatoblastoma, cánceres intestinales, cáncer de estómago y cáncer de tiroides
APC
Dominante
Ataxia-telangiectasia
Leucemia, linfoma
ATM
Recesivo
Síndrome de Beckwith-Wiedemann
Carcinoma suprarrenal, hepatoblastoma, rabdomiosarcoma y tumor de Wilms
CDKN1C, NSD1
Dominante
Síndrome de Bloom
Leucemia, linfoma, cáncer de piel
BLM
Recesivo
Anemia de Diamond-Blackfan
Cáncer de colon, sarcoma osteogénico, LMA/SMD
RPS19 y otros genes RP
Dominante, espontáneob
Anemia de Fanconi
Tumores ginecológicos, leucemia, carcinoma de células escamosas
FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG
Recesivo
Síndrome de poliposis juvenil
Tumores digestivos
SMAD4
Dominante
Síndrome de Li-Fraumeni
Carcinoma corticosuprarrenal, tumor de encéfalo, carcinoma de mama, leucemia, osteosarcoma, sarcoma de tejido blando
TP53
Dominante
Neoplasia endocrina múltiple de tipo 1
Tumor de células de los islotes del páncreas, adenoma de paratiroides, adenoma hipofisario
MEN1
Dominante
Neoplasia endocrina múltiple de tipo 2
Carcinoma medular de tiroides, feocromocitoma
RET
Dominante
Neurofibromatosis de tipo 1
Neurofibroma, glioma de la vía óptica, tumor de vaina del nervio periférico
NF1
Dominante
Neurofibromatosis de tipo 2
Schwannoma vestibular
NF2
Dominante
Síndrome del carcinoma nevoide de células basales
Carcinoma de células basales, meduloblastoma
PTCH
Dominante
Síndrome de Peutz-Jeghers
Cánceres intestinales, carcinoma de ovario, carcinoma de páncreas
STK11
Dominante
Retinoblastoma
Osteosarcoma, retinoblastoma
RB1
Dominante
Esclerosis tuberosa
Hamartoma, angiomiolipoma renal, carcinoma de células renales
TSC1, TSC2
Dominante
Síndrome de Von Hippel-Lindau
Hemangioblastoma, feocromocitoma, carcinoma de células renales, tumores de retina y del sistema nervioso central
VHL
Dominante
Síndrome WAGR
Gonadoblastoma, tumor de Wilms
WT1
Dominante
Síndrome de tumor de Wilms
Tumor de Wilms
WT1
Dominante
Xeroderma pigmentoso
Leucemia, melanoma
XPA, XPB, XPC, XPD, XPE, XPF, XPG, POLH
Recesivo
El instrumento en inglés McGill Interactive Pediatric OncoGenetic Guidelines (MIPOGG) permite identificar a niños con cáncer que tienen mayor probabilidad de presentar un síndrome de predisposición al cáncer. Este instrumento guía a los profesionales clínicos durante una serie de preguntas que se pueden contestar "sí" o "no", y el resultado final indica si se recomienda una evaluación genética.[118,119]
En un estudio de casos y controles anidados en la población, el instrumento MIPOGG identificó a los sobrevivientes con riesgo alto de NMS, después de controlar según la exposición a radiación y quimioterapia (CRI, 1,53; IC 95 %, 1,06–2,19).
La predicción de NMS fue superior en los sobrevivientes de neoplasias en el SNC y tumores sólidos, y en los sobrevivientes no irradiados durante el tratamiento de la neoplasia maligna primaria.
El uso del instrumento MIPOGG en pacientes de oncología pediátrica, en el momento del diagnóstico o durante el seguimiento del sobreviviente, quizás ayude a dar prioridad a las personas que deben someterse a una evaluación genética.
Polimorfismos en enzimas que metabolizan fármacos y que participan en la reparación del DNA
Es más probable que la variabilidad interindividual con respecto al riesgo de NS se relacione con polimorfismos comunes en los genes de penetrancia baja que regulan la disponibilidad de metabolitos activos de fármacos o son responsables de la reparación del DNA. Las interacciones entre los genes y el ambiente quizás magnifiquen las diferencias funcionales sutiles que producen las variaciones genéticas.
En una investigación relacionada, los investigadores del SJLIFE evaluaron los tratamientos del cáncer y las variantes germinales patogénicas en 127 genes de 6 vías de reparación del DNA principales para identificar a los sobrevivientes de cáncer infantil con mayor riesgo de NS.[120]
Entre los 4402 sobrevivientes que se sometieron a secuenciación del genoma completo, 495 (11,2 %) presentaron 1269 NS.
Entre 508 sobrevivientes (11,5 %), se identificaron 538 variantes germinales patogénicas en 98 vías de reparación del DNA (por ejemplo, POLG, MUTYH, ERCC2 y BRCA2).
Se determinó que los siguientes 3 grupos tenían un riesgo elevado de NS:
Las sobrevivientes con variantes de genes de recombinación homóloga que se trataron con dosis altas de radiación (≥20 Gy) dirigida al tórax (CRI, 4,4; IC 95 %, 1,6–12,4) o una dosis acumulada de antraciclinas en el segundo o tercer tercil (CRI, IC 4,4; 95 %, 1,7–11,4) tuvieron un aumento significativo del riesgo de cáncer de mama.
Los sobrevivientes con variantes de genes de recombinación homóloga que recibieron dosis de alquilantes en el tercer tercil tuvieron una mayor tasa de sarcomas subsiguientes (CRI, 14,9; IC 95 %, 4,0–38,0).
Los sobrevivientes con variantes de los genes de reparación por escisión de nucleótidos que se trataron con radiación dirigida al cuello (≥30 Gy) tuvieron un aumento del riesgo de cáncer de tiroides subsiguiente (CRI, 12,9; IC 95 %, 1,6–46,6).
Enzimas que metabolizan fármacos
El metabolismo de los genotóxicos se produce en 2 fases.
La fase l comprende la activación de sustratos en intermediadores electrofílicos de reactividad alta que pueden dañar el DNA, una reacción que lleva a cabo de manera principal la familia de enzimas del citocromo p450 (CYP).
La función de las enzimas de fase II (conjugación) es inactivar los sustratos genotóxicos. Las proteínas de fase II son la glutatión S–transferasa (GST), la NAD(P)H:quinona–oxidorreductasa-1 (NQO1), entre otras enzimas.
El equilibrio entre los 2 grupos de enzimas es de suma importancia para la respuesta celular a los xenobióticos; por ejemplo, la actividad alta de una enzima de fase l y la actividad baja de una enzima de fase II quizás produzcan daños en el DNA.
Polimorfismos de reparación del DNA
Los mecanismos de reparación del DNA protegen las células somáticas de variantes de los genes oncosupresores y los oncogenes que pueden conducir al inicio y progresión del cáncer. La capacidad individual de reparación del DNA está determinada por las características genéticas.[121] Varios genes de reparación del DNA contienen variantes polimórficas que conducen a grandes variaciones interindividuales en la capacidad de reparación del DNA.[121] La evaluación de la contribución de los polimorfismos que influyen en la reparación del DNA en el riesgo de NS es un área activa de investigación.
Riesgo poligénico
Los sobrevivientes de cáncer infantil tienen un mayor riesgo de presentar cánceres posteriores atribuibles a los efectos tardíos de la radioterapia y otras exposiciones al tratamiento. Para comprender mejor cómo afecta la predisposición genética a este riesgo, los cánceres relacionados con la radioterapia y los cánceres relacionados solo con la quimioterapia se han examinado en varios estudios.
Los investigadores combinaron los datos genotípicos de 11 220 sobrevivientes a 5 años del estudio de cohortes CCSS y SJLIFE y llevaron a cabo una investigación exhaustiva de las puntuaciones de riesgo poligénico (PRS) específicas del cáncer en la población general, derivadas de los hallazgos del estudio de asociación del genoma completo. El estudio tenía como objetivo identificar si las PRS estaban relacionadas con el riesgo de cáncer posterior entre los sobrevivientes de cáncer infantil, después de controlar la exposición al tratamiento y los factores de riesgo no genéticos. También cuantificaron las relaciones conjuntas entre la radioterapia y las PRS para comprender la posible interacción entre los factores de riesgo genéticos y el tratamiento.[122]
En los sobrevivientes de ascendencia predominantemente europea, las PRS estandarizadas se relacionaron de forma significativa con el riesgo de CCB posteriores (OR, 1,37; n = 958), cánceres de mama femeninos (OR, 1,42; n = 360), cánceres de tiroides (OR, 1,48; n = 259), carcinomas de células escamosas (OR, 1,20; n = 127) y melanomas (OR, 1,60; n = 103). La relación entre la PRS de cáncer colorrectal y el riesgo posterior de cáncer colorrectal no alcanzó el umbral de significación estadística (OR, 1,19; n = 67).
Los sobrevivientes de cáncer infantil con una PRS más alta que recibieron radioterapia tenían un riesgo más que aditivo de presentar CCB, cánceres de mama y cánceres de tiroides.
En el caso de las mujeres sobrevivientes expuestas a radioterapia torácica (>10 Gy), las que tenían una PRS alta presentaron una mayor incidencia acumulada de cáncer de mama posterior a los 50 años, en comparación con las que tenían una PRS baja.
Con la disminución del uso de la radioterapia, ha sido importante definir la función de la susceptibilidad genética en las NMS relacionadas con la quimioterapia. Los investigadores del estudio de cohortes SJLIFE evaluaron las NMS relacionadas con el tratamiento entre los sobrevivientes a largo plazo de cáncer infantil. Para cada sobreviviente, se calculó una PRS de 179 variantes con validación externa relacionado con el riesgo de cánceres comunes que se presentan en la edad adulta en la población general.[123]
Las NMS más frecuentes que se presentaron en 1594 sobrevivientes incluyeron el CCB (n = 822), el cáncer de mama (n = 235) y el cáncer de tiroides (n = 221).
La relación entre las NMS y las PRS fue mínima en los sobrevivientes de ascendencia europea que recibieron radioterapia (CRI, 1,22; n = 4630).
Entre los sobrevivientes de ascendencia europea que no recibieron radioterapia (n = 4322), el aumento de la incidencia acumulada de NMS a 30 años y el CRI al comparar los quintiles de las PRS superiores e inferiores fue estadísticamente significativo en aquellos que se trataron con alquilantes (17 vs. 6 %; CRI, 2,46; P < 0,01), antraciclinas (20 vs. 8 %; CRI, 2,86; P < 0,001), epipodofilotoxinas (23 vs. 1 %; CRI, 12,20; P < 0,001) o derivados del platino (46 vs. 7 %; CRI, 8,58; P < 0,01).
Entre los sobrevivientes con ascendencia africana que no recibieron radioterapia (n = 414), la PRS también predijo de forma significativa el riesgo de NMS relacionada con las epipodofilotoxinas. Las mayores mejoras en la predicción del riesgo fueron para los sobrevivientes que recibieron epipodofilotoxinas (CRI, 2,68; P < 0,01).
Detección y seguimiento de las neoplasias malignas subsiguientes
Es importante mantener una vigilancia activa para identificar a los sobrevivientes de cáncer infantil en riesgo.[124] Debido al tamaño relativamente pequeño de la población de sobrevivientes de cáncer infantil, así como la prevalencia y el tiempo hasta la aparición de complicaciones relacionadas con el tratamiento, no es factible emprender estudios clínicos para evaluar el efecto de las recomendaciones de detección en la morbilidad y la mortalidad relacionadas con el efecto tardío.
Los estudios bien realizados con poblaciones grandes de sobrevivientes de cáncer infantil aportaron evidencia irrefutable que vincula exposiciones terapéuticas específicas con efectos tardíos. Varios grupos cooperativos nacionales e internacionales (Scottish Collegiate Guidelines Network, Children’s Cancer and Leukaemia Group, Children's Oncology Group [COG], DCOG) usaron esta evidencia para formular por consenso directrices de práctica clínica para aumentar la conciencia y estandarizar las necesidades inmediatas de atención de los sobrevivientes de cáncer infantil vulnerables desde el punto de vista clínico.[125]
En todas las directrices sobre exámenes médicos de detección para los sobrevivientes de cáncer infantil se emplea un abordaje híbrido que se basa, tanto en la evidencia (utilizando relaciones establecidas entre exposiciones terapéuticas y efectos tardíos para identificar categorías de riesgo alto) como en la experiencia clínica colectiva de los expertos (armonizando la magnitud del riesgo con la intensidad de las recomendaciones de detección). Las recomendaciones de detección en estas directrices representan una declaración de consenso de un grupo de expertos en efectos tardíos del tratamiento del cáncer infantil.[124,125]
Las directrices del COG para las NS malignas indican que ciertas poblaciones de riesgo alto entre los sobrevivientes de cáncer infantil, ameritan un enfoque de vigilancia intensificada debido a factores predisponentes del huésped, o factores conductuales y terapéuticos.[124]
Exámenes de detección de la leucemia: el SMD-pCT o la LMA-pCT se suele manifestar dentro de los 10 años posteriores a la exposición. Las recomendaciones incluyen la vigilancia usando los antecedentes y el examen físico en busca de signos y síntomas de pancitopenia durante 10 años después de la exposición a alquilantes o inhibidores de la topoisomerasa II.
Exámenes de detección después de la exposición a la radiación: la mayoría de las otras NS se relacionan con la exposición a la radiación y se suelen manifestar luego de 10 años de la exposición. Las recomendaciones de los exámenes de detección incluyen realizar un cuidadoso examen físico anual de la piel y los tejidos (con frecuencia subyacentes) cercanos al campo de radiación.
Las observaciones específicas para la detección de las NS más comunes relacionados con la radiación son las siguientes:
Exámenes de detección del cáncer de piel de inicio temprano: se recomienda un examen dermatológico anual que se centre en las lesiones cutáneas y los nevos pigmentados en el campo de radiación. Se asesora a los sobrevivientes sobre los siguientes aspectos:
Aumento del riesgo de cáncer de piel.
Posible exacerbación de estos riesgos por el bronceado.
Beneficios de adherirse a comportamientos para proteger la piel de la exposición excesiva a la radiación ultravioleta.
Exámenes de detección del cáncer de mama de inicio temprano: debido a que el desenlace del cáncer de mama se vincula de manera directa con el estadio en el momento del diagnóstico, es posible que la vigilancia minuciosa que permite establecer un diagnóstico temprano confiera una ventaja de supervivencia.[126] Varios grupos de cáncer infantil respaldaron la recomendación de iniciar temprano la vigilancia del cáncer de mama (antes de la detección del cáncer de mama en la población) mediante mamografía, imágenes por resonancia magnética (IRM) de la mama o ambas modalidades de imágenes en mujeres jóvenes que recibieron irradiación dirigida al tórax.[126]
La mamografía, que es el instrumento de detección de aceptación más generalizada para el cáncer de mama en la población general, quizás no sea la herramienta de detección ideal, por sí sola, para cánceres de mama relacionados con la radiación que se presentan en mujeres relativamente jóvenes con mamas densas. De acuerdo con la investigación realizada entre mujeres jóvenes que heredaron la susceptibilidad al cáncer de mama, es posible que los métodos de imágenes dobles mejoren la detección temprana relacionada con una sensibilidad más alta de la IRM para detectar lesiones premenopáusicas en mamas densas y la superioridad de la mamografía para identificar un carcinoma ductal in situ;[127-129] en consecuencia, la American Cancer Society recomienda el uso adicional de la IRM para la detección.[130] La sensibilidad y especificidad altas para detectar las lesiones en estadio temprano de la vigilancia por imágenes dobles se contrarresta con una tasa importante de evaluaciones adicionales atribuibles a resultados positivos falsos.[129]
Muchos médicos se preocupan por los posibles daños vinculados con la exposición a la radiación por la mamografía anual en estas mujeres jóvenes. A este respecto, es importante considerar que se calcula que la media de dosis de radiación dirigida a la mama en las mamografías contemporáneas de detección estándar de 2 proyecciones es de 3,85 a 4,5 mGy.[131-133] Así, 15 mamografías adicionales de detección realizadas entre los 25 y los 39 años aumentarían la exposición total a la radiación en una mujer tratada con 20 Gy de radiación dirigida al del tórax hasta un total de 20,05775 Gy. Los beneficios de la detección de las lesiones tempranas de cáncer de mama en mujeres con riesgo alto se deben equilibrar con la predisposición al riesgo que determina el 0,3 % de exposición adicional a la radiación.
Para mantener a las mujeres jóvenes comprometidas en la vigilancia de la salud de las mamas, en las directrices del COG se recomienda lo siguiente para las mujeres que recibieron una dosis de radiación de 10 Gy o más dirigidas al manto, el mediastino, todo el pulmón y los campos axilares:
Autoexamen mensual de la mama desde la pubertad.
Examen clínico anual de la mama desde la pubertad hasta los 25 años.
Examen clínico de la mama cada 6 meses con mamografía e IRM anuales comenzando 8 años después de la radioterapia o a los 25 años de edad (lo que ocurra más tarde).
El riesgo de cáncer de mama en pacientes que recibieron menos de 10 Gy de radiación con posible efecto en la mama es de una magnitud más baja que para quienes recibieron más de 10 Gy. El control de las pacientes tratadas con menos de 10 Gy de radiación con posible efecto en la mama se determina en forma individual después de una consulta médica sobre los beneficios y riesgos o daños de los exámenes de detección. Si se toma la decisión de administrar los exámenes de detección, se usan las recomendaciones para mujeres expuestas a más de 10 Gy.
Exámenes de detección del cáncer de tiroides: las recomendaciones de vigilancia para el cáncer de tiroides en los sobrevivientes tratados con radioterapia dirigida a la glándula tiroidea varían mucho. El International Guidelines Armonization Group (IGHG) elaboró con el consorcio PanCareSurFup recomendaciones de consenso para la vigilancia. En última instancia, ninguna de las dos estrategias de vigilancia, la ecografía tiroidea y la palpación del cuello, demostró ser superior. Este hallazgo pone de relieve la necesidad de una toma de decisiones compartida entre el proveedor médico y el paciente.[134]
Exámenes de detección de neoplasias subsiguientes en el SNC: en el IGHG se identificó una escasez de evidencia de alta calidad para los exámenes de detección del SNC. El grupo determinó que no había suficiente evidencia para establecer si la detección temprana de neoplasias en el SNC produce una reducción de la morbilidad o la mortalidad. Por lo tanto, no se hizo ninguna recomendación a favor ni en contra de la vigilancia por IRM. Se recomienda la toma de decisiones compartida entre el proveedor médico y la persona sobreviviente para elaborar un plan de detección.[68]
Exámenes de detección del cáncer colorrectal de inicio temprano: los exámenes de detección para quienes están en riesgo de cáncer colorrectal de inicio temprano (es decir, dosis de radiación de 20 Gy o más dirigidas al abdomen, la pelvis o la columna vertebral) incluyen una colonoscopia cada 5 años o una prueba multidirigida de DNA en heces cada 3 años desde los 30 años de edad o 5 años después de la radioterapia (lo que ocurra más tarde). Para obtener más información en inglés, consultar COG Long-Term Follow-Up Guidelines for Survivors of Childhood, Adolescent, and Young Adult Cancers.
Bibliografía
Mertens AC, Liu Q, Neglia JP, et al.: Cause-specific late mortality among 5-year survivors of childhood cancer: the Childhood Cancer Survivor Study. J Natl Cancer Inst 100 (19): 1368-79, 2008. [PUBMED Abstract]
Friedman DL, Whitton J, Leisenring W, et al.: Subsequent neoplasms in 5-year survivors of childhood cancer: the Childhood Cancer Survivor Study. J Natl Cancer Inst 102 (14): 1083-95, 2010. [PUBMED Abstract]
Turcotte LM, Whitton JA, Friedman DL, et al.: Risk of Subsequent Neoplasms During the Fifth and Sixth Decades of Life in the Childhood Cancer Survivor Study Cohort. J Clin Oncol 33 (31): 3568-75, 2015. [PUBMED Abstract]
Turcotte LM, Liu Q, Yasui Y, et al.: Temporal Trends in Treatment and Subsequent Neoplasm Risk Among 5-Year Survivors of Childhood Cancer, 1970-2015. JAMA 317 (8): 814-824, 2017. [PUBMED Abstract]
Stiller CA, Bunch KJ, Bayne AM, et al.: Subsequent cancers within 5 years from initial diagnosis of childhood cancer. Patterns and risks in the population of Great Britain. Pediatr Blood Cancer 70 (5): e30258, 2023. [PUBMED Abstract]
Withrow DR, Anderson H, Armstrong GT, et al.: Pooled Analysis of Meningioma Risk Following Treatment for Childhood Cancer. JAMA Oncol 8 (12): 1756-1764, 2022. [PUBMED Abstract]
Armstrong GT, Liu W, Leisenring W, et al.: Occurrence of multiple subsequent neoplasms in long-term survivors of childhood cancer: a report from the childhood cancer survivor study. J Clin Oncol 29 (22): 3056-64, 2011. [PUBMED Abstract]
van Eggermond AM, Schaapveld M, Lugtenburg PJ, et al.: Risk of multiple primary malignancies following treatment of Hodgkin lymphoma. Blood 124 (3): 319-27; quiz 466, 2014. [PUBMED Abstract]
Bowers DC, Moskowitz CS, Chou JF, et al.: Morbidity and Mortality Associated With Meningioma After Cranial Radiotherapy: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 35 (14): 1570-1576, 2017. [PUBMED Abstract]
Kok JL, Teepen JC, van Leeuwen FE, et al.: Risk of benign meningioma after childhood cancer in the DCOG-LATER cohort: contributions of radiation dose, exposed cranial volume, and age. Neuro Oncol 21 (3): 392-403, 2019. [PUBMED Abstract]
Verbruggen LC, Kok JL, Teepen JC, et al.: Clinical characteristics of subsequent histologically confirmed meningiomas in long-term childhood cancer survivors: A Dutch LATER study. Eur J Cancer 150: 240-249, 2021. [PUBMED Abstract]
Turcotte LM, Liu Q, Yasui Y, et al.: Chemotherapy and Risk of Subsequent Malignant Neoplasms in the Childhood Cancer Survivor Study Cohort. J Clin Oncol 37 (34): 3310-3319, 2019. [PUBMED Abstract]
Teepen JC, van Leeuwen FE, Tissing WJ, et al.: Long-Term Risk of Subsequent Malignant Neoplasms After Treatment of Childhood Cancer in the DCOG LATER Study Cohort: Role of Chemotherapy. J Clin Oncol 35 (20): 2288-2298, 2017. [PUBMED Abstract]
Wang Z, Wilson CL, Easton J, et al.: Genetic Risk for Subsequent Neoplasms Among Long-Term Survivors of Childhood Cancer. J Clin Oncol 36 (20): 2078-2087, 2018. [PUBMED Abstract]
Allodji RS, Hawkins MM, Bright CJ, et al.: Risk of subsequent primary leukaemias among 69,460 five-year survivors of childhood cancer diagnosed from 1940 to 2008 in Europe: A cohort study within PanCareSurFup. Eur J Cancer 117: 71-83, 2019. [PUBMED Abstract]
Allodji RS, Tucker MA, Hawkins MM, et al.: Role of radiotherapy and chemotherapy in the risk of leukemia after childhood cancer: An international pooled analysis. Int J Cancer 148 (9): 2079-2089, 2021. [PUBMED Abstract]
Ghosh T, Hyun G, Dhaduk R, et al.: Late subsequent leukemia after childhood cancer: A report from the Childhood Cancer Survivor Study (CCSS). Cancer Med 13 (20): e70086, 2024. [PUBMED Abstract]
Bhatia S, Yasui Y, Robison LL, et al.: High risk of subsequent neoplasms continues with extended follow-up of childhood Hodgkin's disease: report from the Late Effects Study Group. J Clin Oncol 21 (23): 4386-94, 2003. [PUBMED Abstract]
Nottage K, Lanctot J, Li Z, et al.: Long-term risk for subsequent leukemia after treatment for childhood cancer: a report from the Childhood Cancer Survivor Study. Blood 117 (23): 6315-8, 2011. [PUBMED Abstract]
Khoury JD, Solary E, Abla O, et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 36 (7): 1703-1719, 2022. [PUBMED Abstract]
Thirman MJ, Larson RA: Therapy-related myeloid leukemia. Hematol Oncol Clin North Am 10 (2): 293-320, 1996. [PUBMED Abstract]
Pedersen-Bjergaard J, Philip P: Balanced translocations involving chromosome bands 11q23 and 21q22 are highly characteristic of myelodysplasia and leukemia following therapy with cytostatic agents targeting at DNA-topoisomerase II. Blood 78 (4): 1147-8, 1991. [PUBMED Abstract]
Kok JL, Teepen JC, van der Pal HJ, et al.: Incidence of and Risk Factors for Histologically Confirmed Solid Benign Tumors Among Long-term Survivors of Childhood Cancer. JAMA Oncol 5 (5): 671-680, 2019. [PUBMED Abstract]
Inskip PD, Sigurdson AJ, Veiga L, et al.: Radiation-Related New Primary Solid Cancers in the Childhood Cancer Survivor Study: Comparative Radiation Dose Response and Modification of Treatment Effects. Int J Radiat Oncol Biol Phys 94 (4): 800-7, 2016. [PUBMED Abstract]
Ehrhardt MJ, Howell CR, Hale K, et al.: Subsequent Breast Cancer in Female Childhood Cancer Survivors in the St Jude Lifetime Cohort Study (SJLIFE). J Clin Oncol 37 (19): 1647-1656, 2019. [PUBMED Abstract]
Taylor AJ, Little MP, Winter DL, et al.: Population-based risks of CNS tumors in survivors of childhood cancer: the British Childhood Cancer Survivor Study. J Clin Oncol 28 (36): 5287-93, 2010. [PUBMED Abstract]
Henderson TO, Whitton J, Stovall M, et al.: Secondary sarcomas in childhood cancer survivors: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst 99 (4): 300-8, 2007. [PUBMED Abstract]
Veiga LH, Holmberg E, Anderson H, et al.: Thyroid Cancer after Childhood Exposure to External Radiation: An Updated Pooled Analysis of 12 Studies. Radiat Res 185 (5): 473-84, 2016. [PUBMED Abstract]
Reulen RC, Frobisher C, Winter DL, et al.: Long-term risks of subsequent primary neoplasms among survivors of childhood cancer. JAMA 305 (22): 2311-9, 2011. [PUBMED Abstract]
Teepen JC, Kok JL, Kremer LC, et al.: Long-Term Risk of Skin Cancer Among Childhood Cancer Survivors: A DCOG-LATER Cohort Study. J Natl Cancer Inst 111 (8): 845-853, 2019. [PUBMED Abstract]
Majhail NS, Brazauskas R, Rizzo JD, et al.: Secondary solid cancers after allogeneic hematopoietic cell transplantation using busulfan-cyclophosphamide conditioning. Blood 117 (1): 316-22, 2011. [PUBMED Abstract]
Baker KS, Leisenring WM, Goodman PJ, et al.: Total body irradiation dose and risk of subsequent neoplasms following allogeneic hematopoietic cell transplantation. Blood 133 (26): 2790-2799, 2019. [PUBMED Abstract]
Veiga LH, Curtis RE, Morton LM, et al.: Association of Breast Cancer Risk After Childhood Cancer With Radiation Dose to the Breast and Anthracycline Use: A Report From the Childhood Cancer Survivor Study. JAMA Pediatr 173 (12): 1171-1179, 2019. [PUBMED Abstract]
Kenney LB, Yasui Y, Inskip PD, et al.: Breast cancer after childhood cancer: a report from the Childhood Cancer Survivor Study. Ann Intern Med 141 (8): 590-7, 2004. [PUBMED Abstract]
Moskowitz CS, Chou JF, Wolden SL, et al.: Breast cancer after chest radiation therapy for childhood cancer. J Clin Oncol 32 (21): 2217-23, 2014. [PUBMED Abstract]
Henderson TO, Liu Q, Turcotte LM, et al.: Association of Changes in Cancer Therapy Over 3 Decades With Risk of Subsequent Breast Cancer Among Female Childhood Cancer Survivors: A Report From the Childhood Cancer Survivor Study (CCSS). JAMA Oncol : , 2022. [PUBMED Abstract]
Schaapveld M, Aleman BM, van Eggermond AM, et al.: Second Cancer Risk Up to 40 Years after Treatment for Hodgkin's Lymphoma. N Engl J Med 373 (26): 2499-511, 2015. [PUBMED Abstract]
Travis LB, Hill D, Dores GM, et al.: Cumulative absolute breast cancer risk for young women treated for Hodgkin lymphoma. J Natl Cancer Inst 97 (19): 1428-37, 2005. [PUBMED Abstract]
O'Brien MM, Donaldson SS, Balise RR, et al.: Second malignant neoplasms in survivors of pediatric Hodgkin's lymphoma treated with low-dose radiation and chemotherapy. J Clin Oncol 28 (7): 1232-9, 2010. [PUBMED Abstract]
Inskip PD, Robison LL, Stovall M, et al.: Radiation dose and breast cancer risk in the childhood cancer survivor study. J Clin Oncol 27 (24): 3901-7, 2009. [PUBMED Abstract]
Travis LB, Hill DA, Dores GM, et al.: Breast cancer following radiotherapy and chemotherapy among young women with Hodgkin disease. JAMA 290 (4): 465-75, 2003. [PUBMED Abstract]
van Leeuwen FE, Klokman WJ, Stovall M, et al.: Roles of radiation dose, chemotherapy, and hormonal factors in breast cancer following Hodgkin's disease. J Natl Cancer Inst 95 (13): 971-80, 2003. [PUBMED Abstract]
Lange JM, Takashima JR, Peterson SM, et al.: Breast cancer in female survivors of Wilms tumor: a report from the national Wilms tumor late effects study. Cancer 120 (23): 3722-30, 2014. [PUBMED Abstract]
Wang Y, Ronckers CM, van Leeuwen FE, et al.: Subsequent female breast cancer risk associated with anthracycline chemotherapy for childhood cancer. Nat Med 29 (9): 2268-2277, 2023. [PUBMED Abstract]
Henderson TO, Moskowitz CS, Chou JF, et al.: Breast Cancer Risk in Childhood Cancer Survivors Without a History of Chest Radiotherapy: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 34 (9): 910-8, 2016. [PUBMED Abstract]
Eisenberg ER, Weiss A, Prakash I, et al.: Surgical Management and Contralateral Breast Cancer Risk in Women with History of Radiation Therapy for Hodgkin Lymphoma: Results from a Population-Based Cohort. Ann Surg Oncol 29 (11): 6673-6680, 2022. [PUBMED Abstract]
Moskowitz CS, Ronckers CM, Chou JF, et al.: Development and Validation of a Breast Cancer Risk Prediction Model for Childhood Cancer Survivors Treated With Chest Radiation: A Report From the Childhood Cancer Survivor Study and the Dutch Hodgkin Late Effects and LATER Cohorts. J Clin Oncol 39 (27): 3012-3021, 2021. [PUBMED Abstract]
Dores GM, Anderson WF, Beane Freeman LE, et al.: Risk of breast cancer according to clinicopathologic features among long-term survivors of Hodgkin's lymphoma treated with radiotherapy. Br J Cancer 103 (7): 1081-4, 2010. [PUBMED Abstract]
Horst KC, Hancock SL, Ognibene G, et al.: Histologic subtypes of breast cancer following radiotherapy for Hodgkin lymphoma. Ann Oncol 25 (4): 848-51, 2014. [PUBMED Abstract]
Demoor-Goldschmidt C, Supiot S, Mahé MA, et al.: Clinical and histological features of second breast cancers following radiotherapy for childhood and young adult malignancy. Br J Radiol 91 (1086): 20170824, 2018. [PUBMED Abstract]
Castiglioni F, Terenziani M, Carcangiu ML, et al.: Radiation effects on development of HER2-positive breast carcinomas. Clin Cancer Res 13 (1): 46-51, 2007. [PUBMED Abstract]
Gaffney DK, Hemmersmeier J, Holden J, et al.: Breast cancer after mantle irradiation for Hodgkin's disease: correlation of clinical, pathologic, and molecular features including loss of heterozygosity at BRCA1 and BRCA2. Int J Radiat Oncol Biol Phys 49 (2): 539-46, 2001. [PUBMED Abstract]
Wong SM, Ajjamada L, Weiss AC, et al.: Clinicopathologic features of breast cancers diagnosed in women treated with prior radiation therapy for Hodgkin lymphoma: Results from a population-based cohort. Cancer 128 (7): 1365-1372, 2022. [PUBMED Abstract]
Moskowitz CS, Chou JF, Neglia JP, et al.: Mortality After Breast Cancer Among Survivors of Childhood Cancer: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 37 (24): 2120-2130, 2019. [PUBMED Abstract]
Yeh JM, Lowry KP, Schechter CB, et al.: Clinical Benefits, Harms, and Cost-Effectiveness of Breast Cancer Screening for Survivors of Childhood Cancer Treated With Chest Radiation : A Comparative Modeling Study. Ann Intern Med 173 (5): 331-341, 2020. [PUBMED Abstract]
Vivanco M, Dalle JH, Alberti C, et al.: Malignant and benign thyroid nodules after total body irradiation preceding hematopoietic cell transplantation during childhood. Eur J Endocrinol 167 (2): 225-33, 2012. [PUBMED Abstract]
Friedman DN, Goodman PJ, Leisenring WM, et al.: Impact of risk-based therapy on late morbidity and mortality in neuroblastoma survivors: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst 116 (6): 885-894, 2024. [PUBMED Abstract]
Michaelson EM, Chen YH, Silver B, et al.: Thyroid malignancies in survivors of Hodgkin lymphoma. Int J Radiat Oncol Biol Phys 88 (3): 636-41, 2014. [PUBMED Abstract]
Sigurdson AJ, Ronckers CM, Mertens AC, et al.: Primary thyroid cancer after a first tumour in childhood (the Childhood Cancer Survivor Study): a nested case-control study. Lancet 365 (9476): 2014-23, 2005 Jun 11-17. [PUBMED Abstract]
Clement SC, Lebbink CA, Klein Hesselink MS, et al.: Presentation and outcome of subsequent thyroid cancer among childhood cancer survivors compared to sporadic thyroid cancer: a matched national study. Eur J Endocrinol 183 (2): 169-180, 2020. [PUBMED Abstract]
Casey DL, Vogelius IR, Brodin NP, et al.: Risk of Subsequent Neoplasms in Childhood Cancer Survivors After Radiation Therapy: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 640-654, 2024. [PUBMED Abstract]
Hijiya N, Hudson MM, Lensing S, et al.: Cumulative incidence of secondary neoplasms as a first event after childhood acute lymphoblastic leukemia. JAMA 297 (11): 1207-15, 2007. [PUBMED Abstract]
Neglia JP, Robison LL, Stovall M, et al.: New primary neoplasms of the central nervous system in survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst 98 (21): 1528-37, 2006. [PUBMED Abstract]
Faraci M, Morana G, Bagnasco F, et al.: Magnetic resonance imaging in childhood leukemia survivors treated with cranial radiotherapy: a cross sectional, single center study. Pediatr Blood Cancer 57 (2): 240-6, 2011. [PUBMED Abstract]
Vinchon M, Leblond P, Caron S, et al.: Radiation-induced tumors in children irradiated for brain tumor: a longitudinal study. Childs Nerv Syst 27 (3): 445-53, 2011. [PUBMED Abstract]
Koike T, Yanagimachi N, Ishiguro H, et al.: High incidence of radiation-induced cavernous hemangioma in long-term survivors who underwent hematopoietic stem cell transplantation with radiation therapy during childhood or adolescence. Biol Blood Marrow Transplant 18 (7): 1090-8, 2012. [PUBMED Abstract]
Heymer EJ, Hawkins MM, Winter DL, et al.: Risk of subsequent gliomas and meningiomas among 69,460 5-year survivors of childhood and adolescent cancer in Europe: the PanCareSurFup study. Br J Cancer 130 (6): 976-986, 2024. [PUBMED Abstract]
Bowers DC, Verbruggen LC, Kremer LCM, et al.: Surveillance for subsequent neoplasms of the CNS for childhood, adolescent, and young adult cancer survivors: a systematic review and recommendations from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol 22 (5): e196-e206, 2021. [PUBMED Abstract]
Baliga S, Gallotto S, Bajaj B, et al.: Decade-long disease, secondary malignancy, and brainstem injury outcomes in pediatric and young adult medulloblastoma patients treated with proton radiotherapy. Neuro Oncol 24 (6): 1010-1019, 2022. [PUBMED Abstract]
Wong JR, Morton LM, Tucker MA, et al.: Risk of subsequent malignant neoplasms in long-term hereditary retinoblastoma survivors after chemotherapy and radiotherapy. J Clin Oncol 32 (29): 3284-90, 2014. [PUBMED Abstract]
Fidler MM, Reulen RC, Winter DL, et al.: Risk of Subsequent Bone Cancers Among 69 460 Five-Year Survivors of Childhood and Adolescent Cancer in Europe. J Natl Cancer Inst 110 (2): , 2018. [PUBMED Abstract]
Bright CJ, Hawkins MM, Winter DL, et al.: Risk of Soft-Tissue Sarcoma Among 69 460 Five-Year Survivors of Childhood Cancer in Europe. J Natl Cancer Inst 110 (6): 649-660, 2018. [PUBMED Abstract]
Chaussade A, Millot G, Wells C, et al.: Correlation between RB1germline mutations and second primary malignancies in hereditary retinoblastoma patients treated with external beam radiotherapy. Eur J Med Genet 62 (3): 217-223, 2019. [PUBMED Abstract]
Kube SJ, Blattmann C, Bielack SS, et al.: Secondary malignant neoplasms after bone and soft tissue sarcomas in children, adolescents, and young adults. Cancer 128 (9): 1787-1800, 2022. [PUBMED Abstract]
Schwartz B, Benadjaoud MA, Cléro E, et al.: Risk of second bone sarcoma following childhood cancer: role of radiation therapy treatment. Radiat Environ Biophys 53 (2): 381-90, 2014. [PUBMED Abstract]
Bielack SS, Blattmann C, Hassenpflug W, et al.: Osteosarcoma Arising After Ewing Sarcoma or Vice Versa: A Report of 20 Affected Patients from the Cooperative Osteosarcoma Study Group (COSS). Anticancer Res 43 (11): 4975-4981, 2023. [PUBMED Abstract]
Henderson TO, Rajaraman P, Stovall M, et al.: Risk factors associated with secondary sarcomas in childhood cancer survivors: a report from the childhood cancer survivor study. Int J Radiat Oncol Biol Phys 84 (1): 224-30, 2012. [PUBMED Abstract]
Reulen RC, Winter DL, Diallo I, et al.: Risk Factors for Primary Bone Cancer After Childhood Cancer: A PanCare Childhood and Adolescent Cancer Survivor Care and Follow-Up Studies Nested Case-Control Study. J Clin Oncol 41 (21): 3735-3746, 2023. [PUBMED Abstract]
MacCarthy A, Bayne AM, Brownbill PA, et al.: Second and subsequent tumours among 1927 retinoblastoma patients diagnosed in Britain 1951-2004. Br J Cancer 108 (12): 2455-63, 2013. [PUBMED Abstract]
Temming P, Arendt M, Viehmann A, et al.: Incidence of second cancers after radiotherapy and systemic chemotherapy in heritable retinoblastoma survivors: A report from the German reference center. Pediatr Blood Cancer 64 (1): 71-80, 2017. [PUBMED Abstract]
Kleinerman RA, Schonfeld SJ, Sigel BS, et al.: Bone and Soft-Tissue Sarcoma Risk in Long-Term Survivors of Hereditary Retinoblastoma Treated With Radiation. J Clin Oncol 37 (35): 3436-3445, 2019. [PUBMED Abstract]
Thorsness SL, Freites-Martinez A, Marchetti MA, et al.: Nonmelanoma Skin Cancer in Childhood and Young Adult Cancer Survivors Previously Treated With Radiotherapy. J Natl Compr Canc Netw 17 (3): 237-243, 2019. [PUBMED Abstract]
Boull C, Chen Y, Im C, et al.: Keratinocyte carcinomas in survivors of childhood cancer: A report from the childhood cancer survivor study. J Am Acad Dermatol 91 (6): 1125-1135, 2024. [PUBMED Abstract]
Geller AC, Coroiu A, Keske RR, et al.: Advancing Survivors Knowledge (ASK Study) of Skin Cancer Surveillance After Childhood Cancer: A Randomized Controlled Trial in the Childhood Cancer Survivor Study. J Clin Oncol 41 (12): 2269-2280, 2023. [PUBMED Abstract]
Rotz SJ, Stratton K, Leisenring WM, et al.: Melanoma Among Adult Survivors of Childhood Cancer: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 43 (10): 1219-1228, 2025. [PUBMED Abstract]
Braam KI, Overbeek A, Kaspers GJ, et al.: Malignant melanoma as second malignant neoplasm in long-term childhood cancer survivors: a systematic review. Pediatr Blood Cancer 58 (5): 665-74, 2012. [PUBMED Abstract]
Kleinerman RA, Schonfeld SJ, Abramson DH, et al.: Increased Risk of Skin Cancer in 1,851 Long-Term Retinoblastoma Survivors. J Invest Dermatol 141 (12): 2849-2857.e3, 2021. [PUBMED Abstract]
Ghosh T, Chen Y, Dietz AC, et al.: Lung Cancer as a Subsequent Malignant Neoplasm in Survivors of Childhood Cancer. Cancer Epidemiol Biomarkers Prev 30 (12): 2235-2243, 2021. [PUBMED Abstract]
van Leeuwen FE, Klokman WJ, Stovall M, et al.: Roles of radiotherapy and smoking in lung cancer following Hodgkin's disease. J Natl Cancer Inst 87 (20): 1530-7, 1995. [PUBMED Abstract]
Henderson TO, Oeffinger KC, Whitton J, et al.: Secondary gastrointestinal cancer in childhood cancer survivors: a cohort study. Ann Intern Med 156 (11): 757-66, W-260, 2012. [PUBMED Abstract]
Heymer EJ, Jóźwiak K, Kremer LC, et al.: Cumulative Absolute Risk of Subsequent Colorectal Cancer After Abdominopelvic Radiotherapy Among Childhood Cancer Survivors: A PanCareSurFup Study. J Clin Oncol 42 (3): 336-347, 2024. [PUBMED Abstract]
Tukenova M, Diallo I, Anderson H, et al.: Second malignant neoplasms in digestive organs after childhood cancer: a cohort-nested case-control study. Int J Radiat Oncol Biol Phys 82 (3): e383-90, 2012. [PUBMED Abstract]
Daly PE, Samiee S, Cino M, et al.: High prevalence of adenomatous colorectal polyps in young cancer survivors treated with abdominal radiation therapy: results of a prospective trial. Gut 66 (10): 1797-1801, 2017. [PUBMED Abstract]
Teepen JC, Kok JL, van Leeuwen FE, et al.: Colorectal Adenomas and Cancers After Childhood Cancer Treatment: A DCOG-LATER Record Linkage Study. J Natl Cancer Inst 110 (7): 758-767, 2018. [PUBMED Abstract]
Reulen RC, Wong KF, Bright CJ, et al.: Risk of digestive cancers in a cohort of 69 460 five-year survivors of childhood cancer in Europe: the PanCareSurFup study. Gut : , 2020. [PUBMED Abstract]
Geurts YM, Shakir R, Ntentas G, et al.: Association of Radiation and Procarbazine Dose With Risk of Colorectal Cancer Among Survivors of Hodgkin Lymphoma. JAMA Oncol 9 (4): 481-489, 2023. [PUBMED Abstract]
Sunguc C, Hawkins MM, Winter DL, et al.: Risk of subsequent primary oral cancer in a cohort of 69,460 5-year survivors of childhood and adolescent cancer in Europe: the PanCareSurFup study. Br J Cancer 128 (1): 80-90, 2023. [PUBMED Abstract]
Liu JJ, De Vivo I, Wu CY, et al.: Subsequent primary urogenital cancers among childhood and adolescent cancer survivors in the United States. Urol Oncol 40 (2): 65.e11-65.e18, 2022. [PUBMED Abstract]
Wilson CL, Ness KK, Neglia JP, et al.: Renal carcinoma after childhood cancer: a report from the childhood cancer survivor study. J Natl Cancer Inst 105 (7): 504-8, 2013. [PUBMED Abstract]
de Vathaire F, Scwhartz B, El-Fayech C, et al.: Risk of a Second Kidney Carcinoma Following Childhood Cancer: Role of Chemotherapy and Radiation Dose to Kidneys. J Urol 194 (5): 1390-5, 2015. [PUBMED Abstract]
Hedgepeth RC, Zhou M, Ross J: Rapid development of metastatic Xp11 translocation renal cell carcinoma in a girl treated for neuroblastoma. J Pediatr Hematol Oncol 31 (8): 602-4, 2009. [PUBMED Abstract]
Argani P, Laé M, Ballard ET, et al.: Translocation carcinomas of the kidney after chemotherapy in childhood. J Clin Oncol 24 (10): 1529-34, 2006. [PUBMED Abstract]
Ma J, Pan C, Yin M: Translocation Renal Cell Carcinoma in a Child Previously Treated for Infantile Fibrosarcoma. Pediatr Dev Pathol 21 (4): 418-422, 2018 Jul-Aug. [PUBMED Abstract]
Fleitz JM, Wootton-Gorges SL, Wyatt-Ashmead J, et al.: Renal cell carcinoma in long-term survivors of advanced stage neuroblastoma in early childhood. Pediatr Radiol 33 (8): 540-5, 2003. [PUBMED Abstract]
Henderson TO, Fowler BW, Hamann HA, et al.: Subsequent malignant neoplasms in the Childhood Cancer Survivor Study: Occurrence of cancer types in which human papillomavirus is an established etiologic risk factor. Cancer 128 (2): 373-382, 2022. [PUBMED Abstract]
Ou JY, Bennion N, Parker K, et al.: Risk Factors and Trends for HPV-Associated Subsequent Malignant Neoplasms among Adolescent and Young Adult Cancer Survivors. Cancer Epidemiol Biomarkers Prev 32 (5): 625-633, 2023. [PUBMED Abstract]
Brown AL, Arroyo VM, Agrusa JE, et al.: Survival disparities for second primary malignancies diagnosed among childhood cancer survivors: A population-based assessment. Cancer 125 (20): 3623-3630, 2019. [PUBMED Abstract]
Archer NM, Amorim RP, Naves R, et al.: An Increased Risk of Second Malignant Neoplasms After Rhabdomyosarcoma: Population-Based Evidence for a Cancer Predisposition Syndrome? Pediatr Blood Cancer 63 (2): 196-201, 2016. [PUBMED Abstract]
Wang X, Sun CL, Hageman L, et al.: Clinical and Genetic Risk Prediction of Subsequent CNS Tumors in Survivors of Childhood Cancer: A Report From the COG ALTE03N1 Study. J Clin Oncol 35 (32): 3688-3696, 2017. [PUBMED Abstract]
Waespe N, Belle FN, Redmond S, et al.: Cancer predisposition syndromes as a risk factor for early second primary neoplasms after childhood cancer - A national cohort study. Eur J Cancer 145: 71-80, 2021. [PUBMED Abstract]
Andersson A, Enblad G, Tavelin B, et al.: Family history of cancer as a risk factor for second malignancies after Hodgkin's lymphoma. Br J Cancer 98 (5): 1001-5, 2008. [PUBMED Abstract]
Hisada M, Garber JE, Fung CY, et al.: Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst 90 (8): 606-11, 1998. [PUBMED Abstract]
de Andrade KC, Khincha PP, Hatton JN, et al.: Cancer incidence, patterns, and genotype-phenotype associations in individuals with pathogenic or likely pathogenic germline TP53 variants: an observational cohort study. Lancet Oncol 22 (12): 1787-1798, 2021. [PUBMED Abstract]
Bhatia S, Chen Y, Wong FL, et al.: Subsequent Neoplasms After a Primary Tumor in Individuals With Neurofibromatosis Type 1. J Clin Oncol 37 (32): 3050-3058, 2019. [PUBMED Abstract]
de Blank P, Li N, Fisher MJ, et al.: Late morbidity and mortality in adult survivors of childhood glioma with neurofibromatosis type 1: report from the Childhood Cancer Survivor Study. Genet Med 22 (11): 1794-1802, 2020. [PUBMED Abstract]
Strahm B, Malkin D: Hereditary cancer predisposition in children: genetic basis and clinical implications. Int J Cancer 119 (9): 2001-6, 2006. [PUBMED Abstract]
Cullinan N, Schiller I, Di Giuseppe G, et al.: Utility of a Cancer Predisposition Screening Tool for Predicting Subsequent Malignant Neoplasms in Childhood Cancer Survivors. J Clin Oncol 39 (29): 3207-3216, 2021. [PUBMED Abstract]
Goudie C, Witkowski L, Cullinan N, et al.: Performance of the McGill Interactive Pediatric OncoGenetic Guidelines for Identifying Cancer Predisposition Syndromes. JAMA Oncol 7 (12): 1806-1814, 2021. [PUBMED Abstract]
Qin N, Wang Z, Liu Q, et al.: Pathogenic Germline Mutations in DNA Repair Genes in Combination With Cancer Treatment Exposures and Risk of Subsequent Neoplasms Among Long-Term Survivors of Childhood Cancer. J Clin Oncol 38 (24): 2728-2740, 2020. [PUBMED Abstract]
Collins A, Harrington V: Repair of oxidative DNA damage: assessing its contribution to cancer prevention. Mutagenesis 17 (6): 489-93, 2002. [PUBMED Abstract]
Gibson TM, Karyadi DM, Hartley SW, et al.: Polygenic risk scores, radiation treatment exposures and subsequent cancer risk in childhood cancer survivors. Nat Med 30 (3): 690-698, 2024. [PUBMED Abstract]
Im C, Sharafeldin N, Yuan Y, et al.: Polygenic Risk and Chemotherapy-Related Subsequent Malignancies in Childhood Cancer Survivors: A Childhood Cancer Survivor Study and St Jude Lifetime Cohort Study Report. J Clin Oncol 41 (27): 4381-4393, 2023. [PUBMED Abstract]
Landier W, Bhatia S, Eshelman DA, et al.: Development of risk-based guidelines for pediatric cancer survivors: the Children's Oncology Group Long-Term Follow-Up Guidelines from the Children's Oncology Group Late Effects Committee and Nursing Discipline. J Clin Oncol 22 (24): 4979-90, 2004. [PUBMED Abstract]
Kremer LC, Mulder RL, Oeffinger KC, et al.: A worldwide collaboration to harmonize guidelines for the long-term follow-up of childhood and young adult cancer survivors: a report from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Pediatr Blood Cancer 60 (4): 543-9, 2013. [PUBMED Abstract]
Mulder RL, Hudson MM, Bhatia S, et al.: Updated Breast Cancer Surveillance Recommendations for Female Survivors of Childhood, Adolescent, and Young Adult Cancer From the International Guideline Harmonization Group. J Clin Oncol 38 (35): 4194-4207, 2020. [PUBMED Abstract]
Kriege M, Brekelmans CT, Boetes C, et al.: Efficacy of MRI and mammography for breast-cancer screening in women with a familial or genetic predisposition. N Engl J Med 351 (5): 427-37, 2004. [PUBMED Abstract]
Leach MO, Boggis CR, Dixon AK, et al.: Screening with magnetic resonance imaging and mammography of a UK population at high familial risk of breast cancer: a prospective multicentre cohort study (MARIBS). Lancet 365 (9473): 1769-78, 2005 May 21-27. [PUBMED Abstract]
Tieu MT, Cigsar C, Ahmed S, et al.: Breast cancer detection among young survivors of pediatric Hodgkin lymphoma with screening magnetic resonance imaging. Cancer 120 (16): 2507-13, 2014. [PUBMED Abstract]
Saslow D, Boetes C, Burke W, et al.: American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 57 (2): 75-89, 2007 Mar-Apr. [PUBMED Abstract]
Berrington de Gonzalez A, Berg CD, Visvanathan K, et al.: Estimated risk of radiation-induced breast cancer from mammographic screening for young BRCA mutation carriers. J Natl Cancer Inst 101 (3): 205-9, 2009. [PUBMED Abstract]
Young KC, Burch A, Oduko JM: Radiation doses received in the UK Breast Screening Programme in 2001 and 2002. Br J Radiol 78 (927): 207-18, 2005. [PUBMED Abstract]
Spelic DC: Trends in Mammography Dose and Image Quality 1974-2005. Silver Spring, Md: U.S. Food and Drug Administration, 2006. Available online. Last accessed August 21, 2023.
Clement SC, Kremer LCM, Verburg FA, et al.: Balancing the benefits and harms of thyroid cancer surveillance in survivors of Childhood, adolescent and young adult cancer: Recommendations from the international Late Effects of Childhood Cancer Guideline Harmonization Group in collaboration with the PanCareSurFup Consortium. Cancer Treat Rev 63: 28-39, 2018. [PUBMED Abstract]
Efectos tardíos en el sistema cardiovascular
Se notificó que, después de la recidiva del cáncer primario y la presentación de cánceres secundarios primarios, la enfermedad cardiovascular es la causa principal de mortalidad prematura entre los sobrevivientes a largo plazo de cáncer infantil.[1-3]
Evidencia (exceso de riesgo de mortalidad cardiovascular prematura):
En el North American Childhood Cancer Survivor Study (CCSS) se diagnosticó y trató a más de 34 000 sobrevivientes a 5 años de cáncer infantil entre 1970 y 1999.[1]
Los participantes a los que se les realizó un seguimiento durante 15 años presentaron un razón de mortalidad estandarizada (RME) de 11,7 (intervalo de confianza [IC] 95 %, 9,4–14,4) para la mortalidad cardíaca.
La mortalidad tardía por problemas cardíacos en niños que recibieron tratamiento más recientemente (es decir, en la década de los 90) ha disminuido (por ejemplo, la incidencia acumulada fue del 0,5 % en 1970–1974 vs. el 0,1 % en 1990–1994).
En el CCSS se compararon los desenlaces de los pacientes diagnosticados y tratados en la adolescencia y adultez temprana (de >15 a <21 años) con los sobrevivientes de cánceres similares diagnosticados durante la niñez (de <15 años) y la población general.[4]
Los sobrevivientes de cáncer infantil (n = 5804) presentaron una RME mayor que los sobrevivientes de cáncer diagnosticado durante la adolescencia o adultez temprana (n = 5804) para las muertes cardíacas que comenzaron 20 años después del diagnóstico (RME, 5,4; IC 95 %, 3,7–7,8 vs. RME, 1,8; IC 95 %, 0,7–4,3).
En comparación con hermanos y hermanas de la misma edad, los sobrevivientes de cáncer infantil (n = 4082) tuvieron un riesgo más alto de presentar afecciones cardíacas crónicas de grados 3 a 5 que los casos en adolescentes y jóvenes (n = 4082) (cociente de riesgos instantáneos [CRI], 5,6 [4,5–7,1] vs. 4,3 [3,5–5,4]).
La incidencia y la acumulación de eventos cardiovasculares adversos no graves, incluso las afecciones subclínicas encontradas mediante exámenes de detección, aumentan el riesgo de eventos cardiovasculares adversos graves posteriores en los sobrevivientes de cáncer infantil. En un estudio longitudinal prospectivo de cohortes en el que se usaron evaluaciones clínicas sistemáticas en persona (cohorte St. Jude Lifetime [SJLIFE] formada por 9602 sobrevivientes a 5 años y 737 controles comunitarios) se notificó lo siguiente:[5]
El aumento de la carga acumulada de eventos cardiovasculares adversos no graves de grados 1 a 4 se relacionó con un mayor riesgo futuro de eventos cardiovasculares adversos graves (1 afección: riesgo relativo [RR], 4,3; P < 0,0001; 2 afecciones: RR, 6,6; P < 0,0001; y 3 afecciones: RR, 7,7; P < 0,0001).
Se observó un mayor riesgo de eventos cardiovasculares adversos graves con afecciones subclínicas específicas, como arritmias de grado 1 (RR, 1,5; P = 0,0017), disfunción sistólica ventricular izquierda de grado 2 (RR, 2,2; P < 0,0001) y trastornos valvulares de grado 2 (RR, 2,2; P = 0,013).
El riesgo de eventos cardiovasculares adversos graves no aumentó con la hipercolesterolemia de grado 1, la hipertrigliceridemia de grado 1 a 2 ni con la estenosis vascular de grado 1 a 2.
La enfermedad cardíaca cobra cada vez más importancia a medida que los sobrevivientes de cáncer infantil alcanzan la edad adulta. Este hallazgo se observó en el estudio poblacional British Childhood Cancer Survivor Study, en el que participaron 34 489 sobrevivientes a 5 años de cáncer infantil diagnosticado entre 1940 y 2006.[2,6]
En los sobrevivientes de cáncer infantil de 60 años de edad y más, la enfermedad circulatoria supera a las neoplasias primarias subsiguientes como la causa principal del excedente de mortalidad (el 37 % del número excedente de muertes fue causado por afecciones circulatorias, en comparación con el 31 % de las muertes ocasionadas por neoplasias primarias subsiguientes).[2]
El riesgo de mortalidad cardíaca y por miocardiopatía o insuficiencia cardíaca general fue mayor entre los diagnosticados entre 1980 y 1989. Los sobrevivientes que recibieron el diagnóstico entre 1980 y 1989 tuvieron 28,9 veces el número excedente de muertes por problemas cardíacos que los sobrevivientes que recibieron el diagnóstico ya sea antes de 1970 o de 1990 al presente.[6]
Los efectos tardíos específicos que se tratan en esta sección son los siguientes:
Miocardiopatía e insuficiencia cardíaca.
Cardiopatía isquémica.
Cardiopatía pericárdica.
Valvulopatía.
Trastornos de conducción.
Enfermedad cerebrovascular.
Tromboembolia venoso.
Si bien en esta sección también se tratará con brevedad la influencia de las afecciones relacionadas con estos efectos tardíos, tales como hipertensión, dislipidemia y diabetes, estas afecciones no se analizan en detalle como efecto del tratamiento del cáncer infantil. Se publicó una revisión amplia de la toxicidad cardiovascular a largo plazo en los niños y adultos jóvenes sobrevivientes de cáncer.[7]
Desenlaces cardiovasculares
Varios estudios se centran en los episodios cardíacos entre los sobrevivientes de cáncer infantil. Existen estudios de cohortes muy grandes, muchos con varias décadas de seguimiento, realizados en hospitales,[8-10] con base en ensayos clínicos [11,12] o de carácter poblacional.[2,3,6]
Cabe destacar, que el promedio de edad de estas poblaciones es aún joven (adultos jóvenes o de mediana edad). Por ende, el riesgo notificado de desenlaces cardiovasculares graves quizá sea muy alto en relación con la población general de la misma edad, mientras que el riesgo absoluto a menudo aún es bajo, con lo que limita la potencia de muchos estudios.
Entre los estudios muy grandes con miles de sobrevivientes, la principal limitación ha sido la capacidad inadecuada para determinar de forma clínica las complicaciones cardiovasculares tardías, con una mayor dependencia de los registros administrativos (por ejemplo, registros de defunción), las autonotificaciones o los informes indirectos.
Si bien cada diseño de estudio tiene algunos sesgos inherentes, la bibliografía en general, que se basa en una combinación de desenlaces autonotificados, comprobación clínica y fuentes de datos administrativos, concluye de manera firme que ciertas exposiciones relacionadas con el cáncer predisponen a los sobrevivientes a un riesgo significativamente mayor de morbilidad y mortalidad cardiovasculares.
Aunque la investigación de los efectos tardíos no está al día con los cambios en el tratamiento contemporáneo, muchas terapias vinculadas con los efectos tardíos cardiovasculares aún son de uso común en la actualidad. No obstante, a menudo se administran dosis restringidas de estas terapias a los pacientes con presentaciones de la enfermedad biológicamente favorables. Además, la cardioprotección con dexrazoxano también se usa de manera amplia para los pacientes expuestos a estas terapias.[13-15]
La investigación en curso es importante para asegurar que el uso de los nuevos fármacos dirigidos no produzca efectos cardiovasculares inesperados.[16]
Evidencia (estudios de cohorte seleccionados que describen las tasas de desenlaces cardiovasculares):
En un estudio de casos y controles de la cohorte PanCareSurFup y ProCardio se evaluaron los factores de riesgo de insuficiencia cardíaca relacionados con el tratamiento en sobrevivientes de cáncer infantil (5 o más años) diagnosticados entre 1940 y 2009.[17]
La incidencia acumulada de insuficiencia cardíaca a 50 años en toda la cohorte fue del 2 % (IC 95 %, 1,7–2,2 %).
En un estudio de subcohortes se evaluaron 500 casos de insuficiencia cardíaca sintomática y 500 controles sin insuficiencia cardíaca (emparejados por sexo, edad, año calendario del primer diagnóstico y duración del seguimiento) para quienes se disponía de exposiciones detalladas al tratamiento. El riesgo de insuficiencia cardíaca aumentó más de 5 veces (oportunidad relativa [OR], 5,5; IC 95 %, 2,5–12,3) en los sobrevivientes que recibieron una dosis media de radioterapia cardíaca de 5 a 15 Gy o menos, en comparación con aquellos que no recibieron radioterapia cardíaca. El riesgo aumentó con un volumen más grande de radiación que exponía el corazón.
El riesgo de insuficiencia cardíaca aumentó de manera lineal con dosis medias más altas de radioterapia cardíaca.
Los sobrevivientes que recibieron una dosis acumulada de antraciclina de más de 100 mg/m2 presentaron un aumento sustancial del riesgo de insuficiencia cardíaca (OR, 5,8; IC 95 %, 2,9–11,3 para 100 a <250 mg/m2 y OR, 21,2; IC 95 %, 11,4–39,2 para >250 mg/m2).
Una dosis de antraciclina inferior a 100 mg/m2 no fue un factor de riesgo significativo de insuficiencia cardíaca.
Los investigadores del CCSS notificaron episodios cardíacos de importancia entre los participantes diagnosticados con cáncer infantil entre 1970 y 1999.[18]
En esta actualización, la incidencia acumulada a 20 años de insuficiencia cardíaca y arteriopatía coronaria para pacientes tratados en la década de los 90 disminuyó con el transcurso de las décadas hasta un 0,54 % y un 0,19 %, respectivamente, pero fue significativa solo para la arteriopatía coronaria.
El riesgo de arteriopatía coronaria disminuyó en forma considerable desde las décadas de los 70, 80 y 90 (0,38, 0,24 y 0,19 %, respectivamente; CRI, 0,65) y se atribuyó a reducciones históricas en la exposición a radiación cardíaca, en particular entre sobrevivientes de linfoma de Hodgkin.
Para los pacientes tratados en la década de los 90, la incidencia acumulada a 20 años fue del 0,05 % para la valvulopatía, 0,03 % para la enfermedad pericárdica y 0,13 % para las arritmias; estas cifras no se modificaron con el cambio de época (1970–1990).
En el CCSS, se usaron datos de 24 214 sobrevivientes a 5 años diagnosticados entre 1970 y 1999 para evaluar los efectos de la dosis de radioterapia y el volumen cardíaco expuesto, fármacos quimioterapéuticos específicos y la edad en el momento de la exposición en el riesgo de cardiopatía de inicio tardío.[19]
La incidencia acumulada de enfermedad cardíaca (cualquier cardiopatía, arteriopatía coronaria e insuficiencia cardíaca) 30 años después del diagnóstico fue del 4,8 %. Los varones sobrevivientes tuvieron mayor probabilidad de presentar arteriopatía coronaria y menos probabilidad de presentar insuficiencia cardíaca que las mujeres sobrevivientes. Los sobrevivientes negros que no son hispanos tuvieron mayor probabilidad de presentar cualquier enfermedad cardíaca que los sobrevivientes blancos que no son hispanos.
Las dosis bajas a moderadas de radioterapia (5,0–19,9 Gy) dirigidas a volúmenes cardíacos grandes (>50 % del corazón) se relacionaron con un aumento del riesgo de enfermedad cardíaca de 1,6 veces, en comparación con sobrevivientes que no se expusieron a ninguna radioterapia cardíaca.
Las dosis altas (>20 Gy) dirigidas a volúmenes cardíacos pequeños (0,1–29,9 %) se relacionaron con una tasa elevada de enfermedad cardíaca, en comparación con sobrevivientes no expuestos.
Se observó una relación entre la dosis y la respuesta de la exposición a antraciclina y la insuficiencia cardíaca; los niños más jóvenes (<13 años) presentaron el riesgo más alto de insuficiencia cardíaca después de una dosificación comparable.
En otro estudio del CCSS se evaluaron las relaciones dosis-respuesta de radiación para las subestructuras cardiacas y los desenlaces cardiacos entre 25 481 sobrevivientes a 5 años que recibieron tratamiento entre 1970 y 1999. De estos sobrevivientes, el 48,2 % recibió radioterapia.[20]
Con una mediana de edad de 30 años, la incidencia acumulada (35 años desde el diagnóstico) fue del 3,9 % (IC 95 %, 3,4–4,3 %) para la enfermedad arterial coronaria, del 3,8 % (IC 95 %, 3,4–4,2 %) para la insuficiencia cardiaca, del 1,2 % (IC 95 %, 1,0–1,5 %) para la valvulopatía y del 1,4 % (IC 95 %, 1,1–1,96 %) para la arritmia.
Las dosis medias de 5 a 9,9 Gy dirigidas a todo el corazón no aumentaron el riesgo de ninguna cardiopatía, mientras que las dosis medias de 5 a 9,9 Gy en la arteria coronaria derecha (RR, 2,6; IC 95 %, 1,6–4,1) y en el ventrículo izquierdo (RR, 2,2; IC 95 %, 1,3–3,7) aumentaron el riesgo de enfermedad coronaria.
Las dosis medias de 5 a 9,9 Gy en la válvula tricúspide (RR, 5,5; IC 95 %, 2,0–15,1) y el ventrículo derecho (RR, 8,4; IC 95 %, 3,7–19,0) aumentaron el riesgo de valvulopatía.
El modelado estadístico indicó que cualquier radiación dirigida a las subestructuras cardiacas eleva el riesgo de cardiopatías, y no existe ninguna dosis umbral que mitigue este riesgo.
En un estudio transversal del CCSS participaron 571 adultos sobrevivientes de cáncer infantil en edad adulta (mediana de edad, 37,7 años; 28,5 años desde el diagnóstico del cáncer). En el estudio se compararon las tasas de subdiagnóstico y subtratamiento de los factores de riesgo modificables entre los sobrevivientes y los encuestados de la National Health and Nutrition Examination Survey (NHANES).[21]
Las tasas de subdiagnóstico de la presencia de un factor de riesgo de enfermedad cardiovascular fueron similares (27,1 % de los sobrevivientes vs. 26,1 % de los participantes del NHNES; P = 0,73). Sin embargo, fue más probable que los sobrevivientes se sometieran a tratamiento insuficiente (21,0 versus 13,9 %, P = 0,007; OR, 1,8; IC 95 %, 1,2–2,7).
La hipertensión (18,9 %) y la dislipidemia (16,3 %) representaron los factores de riesgo más subdiagnosticados y subtratados.
Los hombres y los sobrevivientes que tenían sobrepeso u obesidad tenían más probabilidad de recibir un diagnóstico o tratamiento insuficientes.
Estos sobrevivientes de cáncer infantil de edad adulta tuvieron casi el doble de probabilidades de recibir tratamiento insuficiente debido a esas afecciones.
La dislipidemia es más frecuente y constituye un factor predictivo independiente de enfermedad cardiovascular entre los sobrevivientes, en comparación con los controles. Se investigaron anomalías lipídicas específicas y el riesgo de enfermedad cardiovascular aterosclerótica en 4115 sobrevivientes de cáncer infantil de la cohorte SJLIFE.[22]
En esta cohorte, 3406 sobrevivientes no habían recibido ningún diagnóstico previo de dislipidemia.
El colesterol HDL bajo (CRI, 2,9) y los triglicéridos elevados (CRI, 3,1) se relacionaron con un mayor riesgo de infarto de miocardio.
El diagnóstico de colesterol LDL elevado (CRI, 2,2), colesterol no HDL elevado (CRI 2,2), colesterol HDL bajo (CRI, 3,9) y triglicéridos elevados (CRI, 3,8) se relacionó con un mayor riesgo de miocardiopatía.
En un estudio neerlandés del CCSS-LATER, se evaluó la prevalencia y los factores de riesgo de hipertensión en sobrevivientes de cáncer infantil (mediana de edad, 32,5 años; mediana de seguimiento, 25,5 años) que se trataron con terapias potencialmente nefrotóxicas.[23]
Los sobrevivientes (16,3 %) y los controles (18,2 %) presentaron tasas comparables de hipertensión. La hipertensión no diagnosticada prevaleció en el 12 % de los sobrevivientes y el 17,8 % de los controles.
Los factores de tratamiento relacionados con el riesgo de hipertensión fueron la radioterapia abdominal de más de 20 Gy y la irradiación corporal total (ICT).
Entre los sobrevivientes de cáncer infantil, la tasa de filtración glomerular (TFG) inferior a 60 ml/min/1,73 m2 se relacionó de manera significativa con la hipertensión (OR, 3,4; IC 95 %, 1,4–8,5).
El CCSS demostró que la incidencia acumulada de episodios cardíacos graves (infarto de miocardio, insuficiencia cardíaca congestiva, enfermedad pericárdica y anomalías valvulares) en los sobrevivientes de cáncer infantil continúa aumentando más allá de los 45 años de edad.[8]
El riesgo de estos episodios se potenció (es decir, más allá de lo que cabría esperar por un modelo aditivo) por la presencia de afecciones simultáneas posiblemente modificables como obesidad, dislipidemia, diabetes y, en particular, hipertensión.
La hipertensión se relacionó de forma independiente con todos los desenlaces cardíacos graves (razón de tasas, 6 a 19 veces mayor), incluso después del ajuste por el uso de antraciclinas e irradiación dirigida al tórax.
De los 670 sobrevivientes de linfoma de Hodgkin tratados en el St. Jude Children’s Research Hospital (SJCRH) que sobrevivieron 10 años o más, se realizó una evaluación clínica de 348 pacientes en el estudio de cohorte SJLIFE.[24]
En general, los sobrevivientes tenían una carga acumulada más alta (una medición novedosa de la carga de enfermedad en la que se incorporan múltiples afecciones y episodios recidivantes en un solo parámetro) que los controles comunitarios, con la carga total acumulada entre los grados 3 y 5 de los sobrevivientes de 30 años de edad, comparable a la de los controles comunitarios a los 50 años.
En estos sobrevivientes se notificó que, a los 50 años de edad, la incidencia acumulada de por lo menos una afección cardiovascular de grado 3 a grado 5 fue del 45,5 % (IC 95 %, 36,6–54,3 %), en comparación con el 15,7 % (IC 95 %, 7,0–24,4 %) para los controles comunitarios.
El infarto de miocardio y los defectos cardíacos estructurales fueron los que más contribuyeron al exceso de la carga acumulada de grado 3 a grado 5 en los sobrevivientes, mientras que no hubo ninguna diferencia notable entre los sobrevivientes y los controles comunitarios a los 50 años de edad para la carga acumulada de grados 3 a 5 de dislipidemia o hipertensión arterial esencial.
En otro análisis del estudio de cohorte SJLIFE, se comparó la prevalencia de anomalías electrocardiográficas (ECG) graves y leves entre 2715 participantes y 268 controles comunitarios.[25]
Las anomalías graves en el ECG fueron mucho más prevalentes en los sobrevivientes (10,7 %) que en los controles (4,9 %); las anomalías más comunes fueron anomalías aisladas de la onda ST-T (7,2 %), indicios de infarto de miocardio (3,7 %) e hipertrofia ventricular izquierda con rasgos de distensión (2,8 %).
Las exposiciones al tratamiento que predijeron un aumento de riesgo de anomalías graves incluyeron dosis de antraciclina de 300 mg/m2 o más (OR, 1,7; IC 95 %, 1,1–2,5) y radiación cardíaca (OR, 2,1; IC 95 %, 1,5–2,9 [0,01–19,99 cGy]; OR, 2,6; IC 95 %, 1,6–3,9 [20–29,99 cGy]; OR, 10,5; IC 95 %, 6,5–16,9 [≥30 cGy]).
Las anomalías graves en el ECG permitieron pronosticar la mortalidad por todas las causas (CRI, 4,0; IC 95 %, 2,1–7,8).
En el Teenage and Young Adult Cancer Survivor Study, se investigó la mortalidad cardíaca en más de 200 000 sobrevivientes a 5 años de cáncer diagnosticado durante la adolescencia o adultez temprana (edad 15–39 años).[3]
Se determinó que la edad en el momento del diagnóstico y el tipo de cáncer eran importantes para determinar el riesgo de mortalidad cardíaca.
La RME para todas las enfermedades cardíacas combinadas más alta fue la de las personas diagnosticadas entre los 15 y los 19 años (4,2) y disminuyó a 1,2 para aquellas de 35 a 39 años (2P para la tendencia < 0,0001). Este efecto de la edad fue más aparente en los sobrevivientes de linfoma de Hodgkin, en quienes también se concluyó que presentaron el mayor riesgo global.
Las limitaciones de este estudio incluyeron la falta de información detallada sobre las exposiciones a la radioterapia (dosis y campos), las exposiciones a la quimioterapia (en particular, dosis de antraciclina) y los factores de riesgo cardiovascular (por ejemplo, consumo de tabaco, obesidad, hipertensión, diabetes y antecedentes familiares).
Investigadores neerlandeses evaluaron el riesgo de insuficiencia cardíaca, los cambios temporales según los períodos de tratamiento y los factores de riesgo para la insuficiencia cardíaca en 6165 sobrevivientes de cáncer infantil (mediana de edad, 27,3 años; mediana de seguimiento, 19,8 años) diagnosticados entre 1963 y 2002.[26]
La incidencia acumulada de insuficiencia cardíaca 40 años después de un diagnóstico de cáncer infantil fue de un 4,4 % (3,4–5,5 %) entre los miembros de la cohorte.
La incidencia acumulada a 20 años de insuficiencia cardíaca de grado 3 o superior fue más alta en los sobrevivientes que se trataron en los períodos de tratamiento más recientes (para los sobrevivientes diagnosticados entre 1990 y 2001, 1,5 %; entre 1980 y 1989, 1,6 %; y entre 1970 y 1979, 0,5 %) que en los sobrevivientes que se trataron antes; sin embargo, la mortalidad por insuficiencia cardíaca disminuyó en los pacientes que se trataron en períodos más recientes.
Con el análisis multivariante se demostró que los pacientes que recibieron dosis más altas de mitoxantrona o ciclosfosfamida tenían un aumento del riesgo de insuficiencia cardíaca, en comparación con los sobrevivientes que estuvieron expuestos a dosis más bajas.
La iniciativa Pediatric Normal Tissue Effects in the Clinic (PENTEC) informó sobre el riesgo de cardiopatía tardía en sobrevivientes de cáncer infantil tratados con quimioterapia o radioterapia.[27]
Por cada aumento de 10 Gy en la dosis de radiación cardiaca media corregida en fracciones de 1,8 Gy a 2,0 Gy, los CRI estimados fueron de 2,01 (IC 95 %, 1,79–2,25) para la enfermedad arterial coronaria, 1,87 (IC 95 %, 1,70–2,06) para la insuficiencia cardiaca, 1,87 (IC 95 %, 1,78–1,96) para la valvulopatía y 1,88 (IC 95 %, 1,75–2,03) para cualquier cardiopatía.
Por cada aumento de 100 mg/m2 en la dosis acumulada de antraciclina, el CRI fue de 1,93 (IC 95 %, 1,58–2,36) para la aparición de insuficiencia cardiaca, lo que equivale a un aumento en la dosis media de radiación cardiaca de alrededor de 10,5 Gy.
Factores de riesgo del tratamiento
La quimioterapia (en particular las antraciclinas y las antraquinonas) y la radioterapia, administradas de modo independiente o en combinación, aumentan el riesgo de enfermedad cardiovascular de los sobrevivientes de cáncer infantil y se consideran los factores de riesgo más importantes que contribuyen a la enfermedad cardiovascular prematura en esta población.[3,10,17,28]
Antraciclinas y fármacos relacionados
Se sabe que las antraciclinas (por ejemplo, doxorrubicina, daunorrubicina, idarrubicina y epirrubicina) y las antraquinonas (por ejemplo, mitoxantrona) lesionan de forma directa los cardiomiocitos por la inhibición de la topoisomerasa-IIβ en los cardiomiocitos y la formación de especies reactivas al oxígeno, con lo cual se activan vías de muerte celular y la inhibición de la apoptosis mitocondrial.[29,30] Los resultados derivados de la muerte celular son cambios en la estructura del corazón, como el adelgazamiento de la pared, lo que conduce a una sobrecarga ventricular y una remodelación patológica que, con el tiempo, produce disfunción e insuficiencia cardíaca clínica.[31,32]
Los factores de riesgo de la miocardiopatía relacionada con las antraciclinas son los siguientes:[19,33]
Dosis acumulada; en particular, mayor de 250 a 300 mg/m2.
Edad más temprana en el momento de la exposición, en particular, niños menores de 5 años.
Aumento del tiempo transcurrido desde la exposición.
Si bien no hay ningún umbral de dosis baja verdaderamente inocuo, las dosis que superan de 250 a 300 mg/m2 se relacionaron con un aumento importante del riesgo de miocardiopatía, con incidencias acumuladas superiores al 5 % después de 20 años de seguimiento, y en algunos subgrupos, que alcanzan o superan una incidencia acumulada del 10 % a los 40 años de edad.[18,19,32]
La radioterapia simultánea dirigida al tórax o el corazón aumenta aún más el riesgo de miocardiopatía,[10,28,34] así como la presencia de otros rasgos cardiometabólicos como la hipertensión.[8,35]
Si bien la insuficiencia cardíaca clínica a veces ocurre al cabo de unos pocos años de la exposición a la antraciclina, es posible que las manifestaciones clínicas no aparezcan durante décadas en la mayoría de los sobrevivientes, incluso quienes recibieron dosis muy altas.
Equivalencia de las dosis de antraciclinas
En forma tradicional, la equivalencia de las dosis de antraciclinas se ha basado en gran medida en la equivalencia de la toxicidad hematológica aguda, en lugar de la toxicidad cardíaca tardía.
En análisis que reunieron datos agrupados de más de 28 000 sobrevivientes de cáncer infantil a largo plazo, que se siguieron hasta los 40 años de edad (que resultaron en 399 casos de miocardiopatía), se cuestionaron las hipótesis anteriores que consideraban la daunorrubicina equivalente o casi equivalente a la doxorrubicina.[36,37]
En estas investigaciones se encontró que la daunorrubicina era significativamente menos cardiotóxica que la doxorrubicina (relación de equivalencia, 0,5; IC 95 %, 0,4–0,7).[36]
La mitoxantrona, en comparación con la doxorrubicina, es significativamente más cardiotóxica de lo que se pensaba antes (relación de equivalencia, 10,5; IC 95 %, 6,2–19,1), mientras que la epirrubicina fue isoequivalente a la doxorrubicina (relación de equivalencia, 0,8; IC 95 %, 0,3–1,4).[37]
Los datos fueron demasiado escasos para comparar la idarrubicina con la doxorrubicina.
Cardioprotección contra las antraciclinas
Se han explorado estrategias de cardioprotección como las siguientes:
Fármacos y formulaciones liposomales nuevas menos cardiotóxicas. En general, los datos sobre la reducción de la toxicidad de las formulaciones liposomales de antraciclinas en los niños son limitados.[38,39]
Prolongación del tiempo de la infusión. La prolongación del tiempo de infusión se relacionó con una disminución de la insuficiencia cardíaca en pacientes adultos, pero no en niños.[40,41]
Administración simultánea de cardioprotectores. Se han probado diversos fármacos como cardioprotectores (amifostina, acetilcisteína, antagonistas de los canales de calcio, carvedilol, coenzima Q10 y L-carnitina), pero ninguno mostró un beneficio definitivo y no se consideran tratamiento estándar.[42,43]
Dexrazoxano. Hay más datos sobre el dexrazoxano como cardioprotector, pero sobre todo para adultos con cáncer, para quienes la Administración de Alimentos y Medicamentos de los Estados Unidos lo aprobó con el fin de usarlo en mujeres con cáncer de mama metastásico que recibieron 300 mg/m2 de antraciclinas y que tal vez se beneficien con el tratamiento adicional con antraciclinas.[42]
En los datos pediátricos, se observa que el dexrazoxano quizás disminuya algunos marcadores de toxicidad cardíaca temprana hasta 5 años después del tratamiento.[44-47]
Sin embargo, es posible que el dexrazoxano se vincule con un aumento de riesgo de efectos tóxicos agudos en algunos regímenes.[48]
En un estudio a largo plazo se evaluaron los desenlaces de niños con cáncer recién diagnosticados tratados con regímenes con dexrazoxano en ensayos clínicos aleatorizados (dosis acumulada prescrita de doxorrubicina, 100–360 mg/m2; mediana de seguimiento, 18,6 años). El dexrazoxano no se relacionó con recaída del cáncer (CRI, 0,84; IC 95 %, 0,63–1,13), segundos cánceres (CRI, 1,19; IC 95 %, 0,62–2,30), mortalidad por todas las causas (CRI, 1,07; IC 95 %, 0,78–1,47) ni mortalidad cardiovascular (CRI, 1,45; IC 95 %, 0,41–5,16).[49]
En otro estudio se evaluaron los desenlaces cardíacos a largo plazo de 195 niños con leucemia linfoblástica aguda o linfoma de Hodgkin que recibieron doxorrubicina con dexrazoxano o sin este en 4 ensayos aleatorizados y pacientes con osteosarcoma que recibieron doxorrubicina con dexrazoxano en un ensayo no aleatorizado.[50]
A los 18,1 años del diagnóstico de cáncer (51 % expuesto al dexrazoxano; dosis acumulada de doxorrubicina, 297 mg/m2), la administración de dexrazoxano se relacionó con el acortamiento fraccionario del ventrículo izquierdo superior (diferencia absoluta, 1,4 %; IC 95 %, 0,3–2,5) y fracción de eyección (diferencia absoluta, 1,6 %; IC 95 %, 0,0–3,2) y menor estrés miocárdico según el péptido natriurético de tipo B (-6,7 pg/ml; IC 95 %, -10,6 a -2,8).
El uso de dexrazoxano se relacionó con una reducción del riesgo de funcionamiento ventricular izquierdo inferior (acortamiento fraccional del 30 % o fracción de eyección del 50 %; OR, 0,24; IC 95 %, 0,07–0,81); esto se observó sobre todo en aquellos tratados con dosis acumuladas de doxorrubicina de 250 mg/m2 o más.
Radioterapia
Si bien las antraciclinas dañan de forma directa los cardiomiocitos, la radioterapia afecta de manera principal la vasculatura fina de los órganos afectados.[7]
Enfermedad cardiovascular
Los efectos tardíos de la radioterapia dirigida al corazón son específicamente los siguientes:
Pericarditis diferida, que se presenta de forma abrupta o como un derrame pericárdico crónico.
Pancarditis, que incluye fibrosis pericárdica y miocárdica, con fibroelastosis endocárdica o sin esta.
Miocardiopatía (en ausencia de una enfermedad pericárdica importante), que se presenta incluso sin exposición a antraciclinas.
Cardiopatía isquémica.
Lesión funcional en las válvulas, a menudo en las aórticas.
Defectos de conducción.
Estos efectos cardíacos tardíos se relacionan con los siguientes aspectos:
Tamaño de la fracción individual de radiación.
Volumen del corazón expuesto a la radiación.[19,51]
Dosis total de radiación.
En varios estudios se demostró un aumento importante de riesgo de estos desenlaces con dosis más altas de radiación; en particular, dosis dirigidas al corazón que exceden los 35 Gy.[10,12,18,19,28]
Con dosis de radiación más altas, se notificaron tasas superiores al 10 % de insuficiencia cardíaca, enfermedad pericárdica y valvulopatía después de 20 a 30 años. Aunque en algunos estudios se indica que las dosis inferiores a 5 Gy quizás se relacionen con un aumento de riesgo de enfermedad cardiovascular, el riesgo relativo es bajo (es decir, 2,5) y el IC 95 % es alto (es decir, 0,2–41,5); además, los análisis dosimétricos son, por lo general, cálculos de la exposición cardíaca accidental.[10,18,28]
Las dosis bajas a moderadas de radioterapia (5,0–19,9 Gy) dirigidas a volúmenes cardíacos grandes (>50 % del corazón) se relacionan con un aumento del riesgo de enfermedad cardíaca (es decir, 1,6 veces), en comparación con sobrevivientes que no se expusieron a ninguna radioterapia cardíaca.[19]
Las dosis altas de radiación (>20 Gy) dirigidas a volúmenes cardíacos pequeños (0,1–29,9 %) se relacionan con una tasa elevada de enfermedad cardíaca (riesgo relativo, 2,4).[19,52]
En consecuencia, se necesitan datos confirmatorios adicionales para una evaluación exacta del riesgo de dosis cardíacas muy bajas.
De modo similar a las antraciclinas, la manifestación de estos efectos tardíos en ocasiones se demora años y hasta décadas.
Los pacientes expuestos a radioterapia que afecta el sistema cardiovascular y a quimioterapias cardiotóxicas tienen todavía mayor riesgo de desenlaces cardiovasculares tardíos.[10,19] Este riesgo tal vez esté disminuyendo, según los ensayos clínicos de linfoma de Hodgkin del Children's Oncology Group (COG) que abarcan desde 2002 hasta 2022.[53]
En un estudio de cohortes de 2563 pacientes con linfoma de Hodgkin que recibieron tratamiento en cuatro ensayos clínicos consecutivos del COG entre 2002 y 2022 (AHOD0031, AHOD0831, AHOD1331, S1826), se estimó que la incidencia acumulada de cardiopatías de grados 3 a 5 disminuyó, del 10 % en el primer ensayo al 6 % en el último.
Todos los pacientes recibieron doxorrubicina, 1362 recibieron radioterapia mediastínica y 307 recibieron el cardioprotector dexrazoxano (el 80 % de los niños del estudio S1826). La radioterapia se redujo y perfeccionó de forma sustancial, mientras que las dosis de antraciclina aumentaron.
Según la modelización utilizada como parte de este estudio —sobrevivientes tratados en el reciente ensayo de linfoma de Hodgkin de riesgo alto (S1826)— el riesgo a 30 años de morbilidad cardiaca grave aumentará en menos de un 2 % por encima de la tasa esperada en una población no tratada (5 %). Se estimó que las reducciones en la proporción de niños que reciben radioterapia mediastínica y los aumentos en el uso de dexrazoxano compensaron el aumento de la dosis de doxorrubicina, produciendo una reducción neta de las cardiopatías tardías.
Estos resultados indican que se cree que los efectos tóxicos cardiacos serán menos en los ensayos de linfoma de Hodgkin más recientes en comparación con los más antiguos, incluso en mujeres de 12 años o menores, que son el grupo más susceptible a presentar insuficiencia cardiaca inducida por antraciclinas.
Enfermedad cerebrovascular
La enfermedad cerebrovascular después de la exposición a la radioterapia es otro posible efecto tardío que se observa en los sobrevivientes.
El daño vascular inducido por la radiación es un proceso complejo que implica daño tanto arterial como capilar, donde las venas son menos sensibles.
El conjunto de anomalías comprende lesiones lagunares, malformaciones vasculares, telangiectasias, hemorragia intracraneal, moyamoya, microhemorragias y cavernomas, cada una con posibles consecuencias sintomáticas.[54,55]
Mientras que los sobrevivientes de tumores de encéfalo presentan tradicionalmente el mayor riesgo, se notificó que otros sobrevivientes expuestos a irradiación craneal (≥18 Gy) e irradiación dirigida al cuello (≥40 Gy), como los sobrevivientes de leucemia y linfoma, también tienen un aumento de riesgo.[56-59]
En un análisis del PENTEC, se analizó y modeló el riesgo de toxicidad cerebrovascular y el riesgo futuro de accidente cerebrovascular (riesgos proporcionales de COX e incidencias iniciales acumuladas de accidente cerebrovascular en pacientes de cáncer sin radioterapia, con coincidencia de edad y la población general).[60]
De los 3898 pacientes pediátricos analizados en 5 informes, 101 presentaron al menos un efecto tóxico cerebrovascular (por ejemplo, ataque isquémico transitorio [AIT], accidente cerebrovascular, moyamoya o arteriopatía).
El riesgo de cualquier efecto tóxico cerebrovascular fue del 0,2 % a 30 Gy, del 1,3 % a 45 Gy y del 4,4 % a 54 Gy (D50, 75,6 Gy; 68,4–89,4 Gy).
A la edad de 35 años, la incidencia de accidente cerebrovascular prevista a dosis d fue del 0,9 % al 1,3 % a 30 Gy, del 1,8 % al 2,7 % a 45 Gy y del 2,8 % al 4,1 % a 54 Gy (riesgo inicial de la población, 0,2–0,3 %).
A la edad de 45 años, la incidencia de accidente cerebrovascular prevista fue del 2,1 % a 4,2 % a 30 Gy, del 4,5 % al 8,6 % a 45 Gy y del 6,7 % al 13,0 % a 54 Gy (riesgo inicial de la población, 0,5–1,0 %).
Por lo tanto, el riesgo de toxicidad cerebrovascular futura continúa aumentando a medida que aumenta la duración del seguimiento. Además, debido a que el riesgo de accidente cerebrovascular se basó en la dosis prescrita en el lecho tumoral en lugar de la dosis dirigida al polígono de Willis, es probable que se subestimara el riesgo de accidente cerebrovascular notificado.
En los sobrevivientes de linfoma que recibieron solo radioterapia dirigida al tórax o cuello, se cree que la enfermedad cerebrovascular es causada por aterosclerosis de vasos grandes y embolia cardíaca.[57]
El riesgo aumenta con la dosis acumulativa recibida. En un estudio (N = 325) se notificó que el riesgo de accidente cerebrovascular aumentó en un 5 % (CRI, 1,05; IC 95 %, 1,01–1,09; P = 0,02), por cada aumento de 1 Gy en la dosis de radiación, lo que lleva a una incidencia acumulada del 2 % para el primer accidente cerebrovascular después de 5 años y del 4 % después de 10 años.[61]
Los sobrevivientes que sufrieron un accidente cerebrovascular tuvieron un riesgo significativamente mayor de nuevos accidentes cerebrovasculares.[62]
Evidencia (estudios seleccionados en los que se describen la prevalencia y los factores de riesgo de accidente cerebrovascular [ACV] o vasculopatía):
En el estudio poblacional British Childhood Cancer Survivor Study (n = 13 457) se utilizaron datos de Hospital Episode Statistics en Inglaterra para evaluar el riesgo de hospitalizaciones relacionadas con episodios cerebrovasculares (por ejemplo, hemorragia intracraneal no traumática, infarto cerebral u oclusión de la arteria cerebral), en particular en pacientes mayores de 50 años.[59]
Se encontró que un 2,3 % de los sobrevivientes habían sido hospitalizados al menos una vez debido a una enfermedad cerebrovascular, con un riesgo 4 veces mayor, en comparación con la tasa esperada.
Los sobrevivientes de tumores del sistema nervioso central (SNC) o leucemia que recibieron irradiación craneal tuvieron un mayor riesgo de enfermedad cerebrovascular (razón estandarizada de hospitalización [REH] del tumor en el SNC, 15,6; IC 95 %, 14,0–17,4 y leucemia SHR, 5,4; IC 95 %, 4,5–6,4).
Después de los 60 años, en promedio, un 3,1 % de los sobrevivientes de tumores del SNC tratados con irradiación craneal fueron hospitalizados cada año por enfermedad cerebrovascular. A los 65 años, hasta un 26 % de estos pacientes habrán sido hospitalizados debido a un episodio cerebrovascular.
Los investigadores del CCSS observaron que 295 de 13 060 participantes (el 35 % recibió tratamiento con radioterapia craneal) notificaron haber sufrido un accidente cerebrovascular (6,3 %; IC 95 %, 5,1–7,5 %) a los 50 años de edad (mediana de seguimiento, 19 años). Para los sobrevivientes considerados de riesgo alto, la incidencia acumulada de enfermedad cerebrovascular hasta los 50 años fue del 19,9 %.[63]
A partir de la cohorte Euro2K, que incluyó 8 centros de Francia y el Reino Unido, se realizó un estudio retrospectivo de 3172 sobrevivientes de cáncer infantil a 5 años que se vigilaron durante una media de 26 años. Se calcularon las dosis de radiación dirigidas al polígono de Willis recibidas por cada uno de los 2202 niños tratados con radioterapia.[64]
Los pacientes que recibieron radioterapia tuvieron 8,5 veces más riesgo (IC 95 %, 6,3–11,0) de sufrir un accidente cerebrovascular, a diferencia de los pacientes que no recibieron radioterapia, que no tuvieron un riesgo elevado.
El riesgo relativo fue de 15,7 (IC 95 %, 4,9–50,2) con dosis de 40 Gy o más.
A los 45 años de edad, la incidencia acumulada fue del 11,3 % (IC 95 %, 7,1–17,7 %) para los pacientes que recibieron 10 Gy o más dirigidos al polígono de Willis, en comparación con un 1 % para la población general.
Los investigadores del Teenage and Young Adult Cancer Survivor Study (N = 178 962) evaluaron el riesgo de hospitalización por un episodio cerebrovascular entre los sobrevivientes de cáncer a 5 años diagnosticados entre los 15 y 39 años.[65]
Los investigadores encontraron que los sobrevivientes de cánceres diagnosticados durante la juventud y adultez temprana tenían un aumento del 40 % en el riesgo de hospitalización por episodios cerebrovasculares, en comparación con la población general.
Los sobrevivientes de tumores del SNC (SHR, 4,6), tumores de cabeza y cuello (SHR, 2,6) y leucemia (SHR, 2,5) tenían el mayor riesgo de hospitalización por una complicación cerebrovascular.
Los hombres tenían exceso de riesgo absoluto significativamente más altos que las mujeres, en especial, los sobrevivientes de tumores de cabeza y cuello. A los 60 años, el 9 % de los sobrevivientes de tumores del SNC, el 6 % de los sobrevivientes de tumores de cabeza y cuello, y el 5 % de los sobrevivientes de leucemia habían sido hospitalizados por un episodio cerebrovascular.
El riesgo de hospitalización por un infarto cerebral aumentó de manera especial entre los sobrevivientes de un tumor del SNC mayores de 60 años, mientras que este riesgo se incrementó en los sobrevivientes de tumores de cabeza y cuello de todas las edades.
Los investigadores del CCSS evaluaron las tasas y los factores de predicción de accidente cerebrovascular recidivante en los participantes que notificaron un primer accidente cerebrovascular.[62]
Entre los participantes que respondieron (329 de 443), 271 confirmaron un primer accidente cerebrovascular (con una mediana de edad de 19 años) y 70 notificaron un segundo accidente cerebrovascular (con una mediana de edad de 32 años).
Los factores de predicción independientes de accidente cerebrovascular recidivante fueron la radioterapia craneal con una dosis de 50 Gy o más (vs. ausencia de radioterapia craneal), antecedentes de hipertensión y edad de 40 años o más en el momento del primer accidente cerebrovascular (vs. edad 0–17 años).
La incidencia acumulada a 10 años de accidente cerebrovascular tardío recidivante fue del 21 % en general y del 33 % en aquellos participantes tratados con 50 Gy o más de radioterapia craneal.
En un estudio de seguimiento de 224 participantes del CCSS que tuvieron un accidente cerebrovascular se demostró un mayor riesgo de mortalidad por todas las causas y en relación con la salud además de repercusión negativa en el logro social, la función neurocognitiva, el malestar psíquico y otras medidas de la calidad de vida relacionadas con la salud.[66]
En un estudio multicéntrico retrospectivo neerlandés se evaluaron sobrevivientes a 5 años de linfoma de Hodgkin diagnosticado antes de los 51 años (25 % de los pacientes en edad pediátrica) a quienes se sometió a seguimiento durante una mediana de 18 años. Entre 2201 sobrevivientes, 96 presentaron enfermedad cerebrovascular (ACV y AIT).[57]
La mayoría de los episodios isquémicos fueron por aterosclerosis en una arteria grande (36 %) o embolia cardíaca (24 %).
La incidencia acumulada de ACV isquémico o AIT 30 años después del tratamiento del linfoma fue del 7 %.
El razón de incidencia estandarizada (RIE) para ACV fue de 2,2 y de 3,1 para AIT. Sin embargo, los cálculos de la RIE fueron mayores en los sobrevivientes de cáncer infantil, con un CIE de 3,8 para ACV y de 7,6 para AIT.
La irradiación dirigida al cuello y el mediastino fue un factor de riesgo independiente de enfermedad cerebrovascular isquémica (CRI, 2,5; IC 95 %, 1,1–5,6) versus la ausencia de radioterapia. El tratamiento con quimioterapia no se relacionó con un aumento del riesgo.
La hipertensión, la diabetes mellitus y la hipercolesterolemia se relacionaron con la presentación de enfermedad cerebrovascular isquémica.
Tromboembolismo venoso
Los niños con cáncer tienen un exceso de riesgo de tromboembolismo venoso dentro de los primeros 5 años posteriores al diagnóstico; sin embargo, no se ha estudiado el riesgo a largo plazo de tromboembolismo venoso entre los sobrevivientes de cáncer infantil.[67]
Los investigadores del CCSS evaluaron el tromboembolismo venoso autonotificado de inicio tardío (5 años o más después del diagnóstico de cáncer) entre los miembros de la cohorte (mediana de seguimiento de 21,3 años).[68]
La incidencia acumulada a 35 años de tromboembolismo venoso en los sobrevivientes fue del 4,9 %, lo cual representa un riesgo más de 2 veces superior, en comparación con la cohorte de hermanos (razón de tasas, 2,2; IC 95 %, 1,7–2,8).
Los factores de riesgo de tromboembolismo venoso en los sobrevivientes incluyeron el sexo femenino, el tratamiento con cisplatino o asparaginasa, la obesidad o el peso insuficiente, un cáncer primario recidivante o un cáncer subsiguiente.
El riesgo de tromboembolismo venoso tardío fue más alto en los sobrevivientes de osteosarcoma de extremidad inferior tratados con cirugía con conservación del miembro, en comparación con los pacientes tratados con amputación, quizás por las alteraciones en la anatomía vascular periférica y en la homeostasis.
El tromboembolismo venoso se relacionó con casi el doble de riesgo de mortalidad tardía (RR, 1,9; IC 95 %, 1,6–2,3).
Afecciones cardiovasculares convencionales
Hay diversas exposiciones a tratamientos del cáncer que influyen también directa o indirectamente en la presentación de hipertensión, diabetes mellitus y dislipidemia.
Estas afecciones son tan importantes para los sobrevivientes de cáncer como para la población general porque son factores de riesgo independientes para la presentación de miocardiopatía, cardiopatía isquémica y enfermedad cerebrovascular.[8,69-72]
Los sobrevivientes de cáncer infantil se deben someter a seguimiento meticuloso para detectar estas afecciones porque representan objetivos de intervención potencialmente modificables.
Esto incluye la vigilancia de las afecciones relacionadas, como la obesidad y diversas endocrinopatías (por ejemplo, hipotiroidismo, hipogonadismo y deficiencia de la hormona del crecimiento), que son más comunes en subgrupos de sobrevivientes de cáncer infantil; si estas afecciones no se tratan o controlan, es posible que se vinculen con un perfil metabólico que aumenta el riesgo cardiovascular.[9] Para obtener más información, consultar la sección Predicción del riesgo e intervenciones en enfermedades cardiovasculares.
Otros factores de riesgo
Sexo. En algunos estudios, aunque no en todos, el sexo femenino se relaciona con un riesgo mayor de miocardiopatía relacionada con las antraciclinas.[7]
Características genéticas. La evidencia emergente indica que los factores genéticos, como los polimorfismos de un solo nucleótido en los genes que regulan el metabolismo y la distribución de los fármacos, podrían explicar la heterogeneidad de la susceptibilidad a lesiones cardíacas mediadas por antraciclinas.[73-81] Sin embargo, estos hallazgos genéticos aún necesitan validación adicional antes de incorporarse en cualquier algoritmo de detección clínica.[81]
Disfunción cardíaca del periparto
Los sobrevivientes a largo plazo de neoplasias malignas diagnosticadas durante la niñez, adolescencia y adultez temprana con exposición anterior a tratamientos potencialmente cardiotóxicos están en riesgo de disfunción cardíaca del periparto.
En la población general, la miocardiopatía del periparto o puerperal es una afección inusual caracterizada por la insuficiencia cardíaca durante el embarazo (por lo general en el último trimestre o <5 meses del período puerperal). La incidencia estimada en la población general es de 1 caso por 3000 recién nacidos vivos.[82]
Los datos sobre la prevalencia en sobrevivientes de neoplasias malignas diagnosticadas durante la niñez, la adolescencia y la adultez temprana que recibieron tratamientos cardiotóxicos son limitados.
En una serie retrospectiva del SJCRH, se produjeron 3 casos de disfunción cardíaca del periparto en 1554 embarazos completados, que corresponde a una incidencia del 0,2 %; el 27 % de las 847 sobrevivientes a largo plazo no se habían expuesto a tratamientos cardiotóxicos.[83]
En una serie de 64 mujeres que habían recibido tratamiento cardiotóxico (el 44 % recibió radioterapia dirigida al tórax y antraciclinas, 14 % recibió radioterapia dirigida al tórax, el 42 % recibió solo antraciclinas y 5 mujeres (7,8 %) tuvieron episodios cardíacos durante el periparto (3 sintomáticos, 2 subclínicos). De los 110 recién nacidos vivos, 2 madres se clasificaron con diagnóstico de miocardiopatía del periparto, lo cual representa un riesgo 55 veces mayor que la población general. Los factores de riesgo fueron la edad más joven en el momento del diagnóstico de cáncer y una dosis de antraciclinas más alta. En 4 mujeres, la función cardíaca en el posparto no volvió al estado inicial (80 %).[84]
En un estudio de un solo centro se calculó el riesgo de presentar insuficiencia cardíaca congestiva (ICC) durante el embarazo en 1 de 3 mujeres sobrevivientes de cáncer con antecedentes de cardiotoxicidad.[85] Se identificaron retrospectivamente 78 mujeres sobrevivientes consecutivas de cáncer expuestas con anterioridad a tratamientos con potencial cardiotóxico (quimioterapia o radioterapia dirigida al tórax en niños, adolescentes o adultos jóvenes) que tuvieron 94 embarazos en clínicas de riesgo alto entre 2005 y 2015.
Un total de 55 mujeres recibieron antraciclinas (intervalo, 90–500 mg/m2), mientras que 23 recibieron quimioterapia sin antraciclinas o radioterapia solamente. De las 13 mujeres con cardiotoxicidad previa, 8 tenían una fracción de eyección ventricular izquierda reducida en la primera consulta prenatal. La ICC se presentó en 5 embarazos (4 mujeres; 5,3 %).
La incidencia de ICC fue del 31 % en las mujeres con antecedentes de cardiotoxicidad y del 0 % en las mujeres sin dichos antecedentes. Cuando se compararon las características clínicas de las mujeres con ICC o sin ICC, no hubo ninguna diferencia en la edad del diagnóstico de cáncer, el tipo de cáncer o la exposición a las antraciclinas.
Los factores de riesgo de presentar ICC fueron antecedentes de cardiotoxicidad antes del embarazo, disfunción sistólica del ventrículo izquierdo en la consulta visita prenatal o recibir medicamentos para el corazón.
A partir de la evidencia disponible sobre la miocardiopatía periparto, el International Guideline Armonization Group consideró que la vigilancia de la miocardiopatía es razonable antes del embarazo o en el primer trimestre para las mujeres sobrevivientes de cáncer diagnosticado durante la niñez, adolescencia y adultez temprana con riesgo moderado y alto porque se trataron con antraciclinas o radioterapia torácica.[33]
Riesgo de mortalidad después de episodios cardiovasculares graves
Los sobrevivientes de cáncer infantil representan una población de riesgo alto de mortalidad tras episodios cardiovasculares graves. Los investigadores calcularon la incidencia acumulada de mortalidad por todas las causas, así como por causas cardiovasculares específicas, entre los sobrevivientes del CCSS que habían sufrido un episodio cardiovascular grave y las compararon con las de sus hermanos. También compararon los resultados de los sobrevivientes de cáncer del CCSS con una cohorte poblacional de adultos de diversas razas del estudio Coronary Artery Risk Development in Young Adults (CARDIA).[86]
Entre los 25 658 sobrevivientes de cáncer infantil (mediana de edad en el momento del diagnóstico, 7 años; mediana de edad en el momento del seguimiento o la muerte, 38 años) y 5051 hermanos, 1780 sobrevivientes y 91 hermanos experimentaron un episodio cardiovascular.
La tasa de mortalidad por todas las causas a 10 años para los sobrevivientes fue del 30 % tras insuficiencia cardiaca, del 36 % tras enfermedad arterial cardiovascular y del 29 % después de un accidente cerebrovascular. La tasa de mortalidad por todas las causas a los 10 años para los hermanos fue del 14 % después de insuficiencia cardiaca, del 14 % tras enfermedad arterial coronaria y del 4 % después de accidente cerebrovascular (P < 0,001 para todos los sobrevivientes).
Los riesgos de mortalidad por todas las causas entre los sobrevivientes de cáncer infantil aumentaron después de insuficiencia cardiaca (CRI, 7,32), enfermedad arterial coronaria (CRI, 5,54) y accidente cerebrovascular (CRI, 3,57).
De los 5114 participantes en el estudio CARDIA, 345 sufrieron un episodio grave. Los participantes en CARDIA eran, en promedio, décadas mayores en el momento de los episodios (mediana de edad, 57 años vs. 31 años). Sin embargo, los riesgos de mortalidad fueron similares, salvo que la mortalidad por todas las causas tras enfermedad coronaria aumentó significativamente entre los sobrevivientes de cáncer infantil (CRI, 1,85).
Trasplante de corazón después de cáncer infantil
Los datos sobre la prevalencia y los desenlaces de los sobrevivientes con insuficiencia cardíaca que necesitan trasplante de corazón son limitados.
En un estudio de trasplantes de vísceras macizas con 13 318 sobrevivientes en el CCSS, 62 sobrevivientes presentaban insuficiencia cardíaca en etapa terminal que justificaba trasplante de corazón y 37 de ellos recibieron dicho trasplante.[87]
A los 35 años del diagnóstico de cáncer, la incidencia acumulada del trasplante de corazón fue del 0,30 %, y la incidencia acumulada de entrar en la lista de espera o de recibir un corazón fue del 0,49 %.[87]
La tasa de supervivencia a 5 años del trasplante de corazón fue del 80,6 %, que es similar al desenlace en la población general del mismo intervalo de edad.[87]
Conocimientos insuficientes
A pesar del abundante conocimiento adquirido en los últimos 20 años para comprender mejor la carga y los factores de riesgo a largo plazo de la enfermedad cardiovascular en los sobrevivientes de cáncer infantil, aún se necesita investigar muchas áreas como las siguientes:
Es posible que la radiación tenga efectos directos e indirectos en el endotelio vascular, lo que contribuye al daño vascular más allá del campo primario de radiación.[88]
Aún se deben determinar los efectos a largo plazo de las dosis de radiación más bajas, en particular en el entorno de tecnología avanzada que permite centrarse en el tumor desde múltiples direcciones y reduce la exposición de tejidos normales adyacentes.[89]
Todavía no están claros los efectos a largo plazo de los antineoplásicos más nuevos que se basan en objetivos moleculares, aunque se sabe que algunos de ellos producen efectos tóxicos cardíacos en el corto plazo.[16]
Es necesario estudiar más la eficacia de las estrategias de protección cardíaca en los niños, incluso el uso de formulaciones alternativas de antraciclinas que han resultado alentadoras para los adultos.[43]
Exámenes de detección, vigilancia y asesoramiento
El International Guideline Armonization Group ha trabajado de manera colaborativa para armonizar las recomendaciones de vigilancia cardíaca basadas en evidencia y ha identificado carencias de conocimiento para ayudar a orientar los estudios futuros.[33,72] Los grupos de riesgo, definidos por exposiciones acumuladas de antraciclina y radioterapia dirigida al tórax, así como las recomendaciones de vigilancia de la miocardiopatía se resumen en el Cuadro 2.
Consenso en torno a exámenes de detección, vigilancia y asesoramiento
No hay evidencia clara (al menos hasta los 50 años de edad o 30 a 40 años después del tratamiento) de que se produzca una meseta en el riesgo después de cierto tiempo para los sobrevivientes expuestos a tratamientos de cáncer relacionados con efectos tardíos cardiovasculares.[90,91] Por lo tanto, se recomienda vigilancia de por vida para uno de los grupos, aunque la relación de la eficacia en función del costo de ciertas estrategias de detección todavía no está clara.[33,92-94]
Cuadro 2. Recomendaciones sobre los grupos de riesgo y la vigilancia de la miocardiopatía para sobrevivientes de cáncer diagnosticado durante la niñez, adolescencia y adultez tempranaa
Grupo de riesgo
Antraciclina (mg/m2)
Radioterapia dirigida al tórax (Gy)
Antraciclina (mg/m2) y radioterapia dirigida al tórax (Gy)
Cada vez hay más bibliografía en la que se comienza a establecer el rendimiento de estos estudios de detección, lo cual ayudará a guiar la formulación de directrices futuras.[9,95-97] Por ejemplo, en estos estudios de adultos sobrevivientes de cáncer infantil, se encontraron indicios de miocardiopatía de acuerdo con cambios ecocardiográficos en casi el 6 % de los sobrevivientes en riesgo. En general, en una cohorte de más de 1000 sobrevivientes (mediana de edad, 32 años), en casi un 60 % de los sobrevivientes en riesgo sometidos a detección se identificó alguna anomalía cardíaca mediante comprobación clínica.[9]
Dado el aumento de evidencia que avala que las afecciones cardiovasculares convencionales, como hipertensión, dislipidemia y diabetes, aumentan mucho el riesgo de enfermedades cardiovasculares más graves entre los sobrevivientes, los médicos deben considerar con cuidado los valores de referencia y los exámenes de detección de seguimiento, así como el tratamiento de estas afecciones comórbidas que afectan la salud cardiovascular (consultar el Cuadro 3).[8,57,69,98]
También hay nueva evidencia que indica que la adopción de factores de estilo de vida más sanos disminuye la morbilidad cardiovascular futura en sobrevivientes en riesgo.[99,100] Por lo tanto, se asesora a los sobrevivientes, igual que a la población general, para que mantengan un peso saludable, participen con regularidad en actividades físicas, cumplan con un régimen de alimentación saludable para el corazón y se abstengan de fumar.
El COG ha elaborado folletos sobre la enfermedad cardiovascular y temas conexos, incluso opciones para un estilo de vida saludable escritos en términos coloquiales para facilitar el asesoramiento y la información para los sobrevivientes. Para obtener más información en inglés, consultar COG Survivorship Guidelines.
Predicción del riesgo de enfermedades cardiovasculares
Los intentos por formular una predicción más individualizada del riesgo de enfermedad cardiovascular podrían contribuir a refinar la vigilancia y el asesoramiento en el futuro.
Varios grupos han colaborado para formular y validar calculadoras de riesgo individualizado para insuficiencia cardíaca, enfermedad cardíaca isquémica y accidente cerebrovascular hasta los 50 años de edad.[34,63,98]
Modelos actualizados basados solo en datos del CCSS han incorporado la hipertensión, la dislipidemia y la diabetes, además de su evolución temporal para refinar más la predicción.[98]
Una calculadora del riesgo en línea que incorpora estos modelos se puede consultar en el siguiente enlace en inglés del sitio web del CCSS.
Predicción del riesgo e intervenciones en enfermedades cardiovasculares
Se ha creado y validado una calculadora de riesgo de insuficiencia cardíaca sobre la base de las características demográficas y de tratamiento fácilmente disponibles, con datos de 4 cohortes grandes de sobrevivientes de cáncer infantil bien anotadas (CCSS, National Wilms Tumor Study Group, los Países Bajos y SJCRH) que es posible que proporcione un cálculo más individualizado del riesgo de insuficiencia cardíaca clínica de los sobrevivientes a 5 años de cáncer infantil que recién completaron el tratamiento y hasta los 40 años de edad. Debido a la edad temprana de los participantes en el momento de la predicción inicial (supervivencia a 5 años), este estimador es limitado, dado que no se pudo incorporar información sobre afecciones cardiovasculares convencionales, tales como hipertensión, dislipidemia o diabetes.[34]
En otro estudio de colaboración, se usaron datos del CCSS, los Países Bajos y el SJCRH para formular modelos de predicción del riesgo a 50 años de cardiopatía isquémica y accidente cerebrovascular en sobrevivientes de cáncer infantil a 5 años. Mediante el empleo de puntuaciones de riesgo derivadas de un modelo de predicción estándar que incluía sexo, exposición a la quimioterapia y exposición a la radioterapia, se identificaron grupos diferenciados a partir del análisis estadístico: riesgo bajo, riesgo moderado y riesgo alto. Las incidencias acumuladas a los 50 años en los grupos de riesgo bajo del CCSS fueron inferiores al 5 %, en comparación con cerca del 20 % en los grupos de riesgo alto y solo el 1 % en los hermanos o hermanas.[63]
Los factores de riesgo cardiovascular tradicionales siguen siendo importantes para predecir el riesgo de enfermedad cardiovascular entre los adultos sobrevivientes de cáncer infantil, como se demostró en una investigación del CCSS que construyó modelos de predicción teniendo en cuenta las exposiciones a tratamientos del cáncer cardiotóxicos, en combinación con información sobre los factores de riesgo cardiovascular tradicionales como la hipertensión, la dislipidemia y la diabetes. Las puntuaciones de riesgo a partir información demográfica, del tratamiento del cáncer, hipertensión, dislipidemia y diabetes demostraron un buen desempeño (área bajo la curva de rendimiento diagnóstico y el estadístico de concordancia ≥0,70) para predecir episodios cardiovasculares en los modelos aplicados a cohortes de descubrimiento y replicación. Las exposiciones con mayor influencia fueron el uso de antraciclinas, el uso de radioterapia, la presencia de diabetes y la presencia de hipertensión.[98]
Se ha demostrado que la actividad física es una intervención inocua contra la gran carga de efectos tardíos cardiovasculares en sobrevivientes de cáncer infantil. En un ensayo controlado y aleatorizado se investigó el efecto de una intervención de actividad física supervisada de manera parcial, personalizada, de 1 año de duración, sobre la salud cardiovascular en sobrevivientes a largo plazo de cáncer infantil.[101] Se asignó al azar a 151 sobrevivientes de cáncer infantil (1:1) a realizar más de 2,5 horas de actividad física intensa adicional por semana (grupo de intervención) o continuar realizando el ejercicio físico habitual (grupo de control). Se observó una reducción significativa y consistente de la puntuación de riesgo de enfermedad cardiovascular a los 6 y 12 meses en el grupo de intervención, en comparación con el grupo de control. La diferencia en la reducción de la puntuación z del riesgo de enfermedad cardiovascular de -0,18 (P = 0,003) a los 12 meses favoreció al grupo de intervención.
Cuadro 3. Efectos tardíos cardiovascularesa,b
Tratamiento predisponente
Posibles efectos cardiovasculares
Exámenes médicos de detección
TCMH = trasplante de células madre hematopoyéticas.
aLas directrices del Children's Oncology Group (COG) también se refieren a otras afecciones que tal vez repercutan en el riesgo cardiovascular, como obesidad y diabetes mellitus o deterioro del metabolismo de la glucosa.
Cualquier antraciclina o radiación dirigida al corazón
Toxicidad cardíaca (arritmia, miocardiopatía o insuficiencia cardíaca, enfermedad pericárdica, valvulopatía y cardiopatía isquémica)
Antecedentes médicos y examen físico anuales
Electrocardiograma al principio del seguimiento a largo plazo
Ecocardiograma al principio del seguimiento a largo plazo, repetido de forma periódica de acuerdo con las exposiciones previas y otros factores de riesgo
Radiación dirigida al cuello y la base del cráneo (en especial, ≥40 Gy)
Arteriopatía carotídea o subclavia
Antecedentes médicos y examen físico anuales; considerar una ecografía Doppler 10 años después de la exposición
Radiación dirigida al encéfalo o el cráneo (en especial, ≥18 Gy)
Enfermedad cerebrovascular (cavernomas, moyamoya, vasculopatía cerebral oclusiva y accidente cerebrovascular)
Antecedentes médicos y examen físico anuales
Radiación dirigida al abdomen
Diabetes
Exámenes de detección de diabetes cada 2 años
Irradiación corporal total (por lo general <14 Gy)
Dislipidemia; diabetes
Lipidograma en ayunas y exámenes de detección de diabetes cada 2 años
Exposición a metales pesados (carboplatino y cisplatino) e ifosfamida; radiación dirigida a los riñones; TCMH y nefrectomía
Hipertensión (debido a toxicidad renal)
Evaluaciones anuales de la presión arterial; estudios de laboratorio del funcionamiento renal al principio del seguimiento a largo plazo y repetición conforme se indique clínicamente.
Bibliografía
Armstrong GT, Chen Y, Yasui Y, et al.: Reduction in Late Mortality among 5-Year Survivors of Childhood Cancer. N Engl J Med 374 (9): 833-42, 2016. [PUBMED Abstract]
Fidler MM, Reulen RC, Winter DL, et al.: Long term cause specific mortality among 34 489 five year survivors of childhood cancer in Great Britain: population based cohort study. BMJ 354: i4351, 2016. [PUBMED Abstract]
Henson KE, Reulen RC, Winter DL, et al.: Cardiac Mortality Among 200 000 Five-Year Survivors of Cancer Diagnosed at 15 to 39 Years of Age: The Teenage and Young Adult Cancer Survivor Study. Circulation 134 (20): 1519-1531, 2016. [PUBMED Abstract]
Suh E, Stratton KL, Leisenring WM, et al.: Late mortality and chronic health conditions in long-term survivors of early-adolescent and young adult cancers: a retrospective cohort analysis from the Childhood Cancer Survivor Study. Lancet Oncol 21 (3): 421-435, 2020. [PUBMED Abstract]
Hammoud RA, Liu Q, Dixon SB, et al.: The burden of cardiovascular disease and risk for subsequent major adverse cardiovascular events in survivors of childhood cancer: a prospective, longitudinal analysis from the St Jude Lifetime Cohort Study. Lancet Oncol 25 (6): 811-822, 2024. [PUBMED Abstract]
Fidler MM, Reulen RC, Henson K, et al.: Population-Based Long-Term Cardiac-Specific Mortality Among 34 489 Five-Year Survivors of Childhood Cancer in Great Britain. Circulation 135 (10): 951-963, 2017. [PUBMED Abstract]
Bansal N, Blanco JG, Sharma UC, et al.: Cardiovascular diseases in survivors of childhood cancer. Cancer Metastasis Rev 39 (1): 55-68, 2020. [PUBMED Abstract]
Armstrong GT, Oeffinger KC, Chen Y, et al.: Modifiable risk factors and major cardiac events among adult survivors of childhood cancer. J Clin Oncol 31 (29): 3673-80, 2013. [PUBMED Abstract]
Hudson MM, Ness KK, Gurney JG, et al.: Clinical ascertainment of health outcomes among adults treated for childhood cancer. JAMA 309 (22): 2371-81, 2013. [PUBMED Abstract]
Haddy N, Diallo S, El-Fayech C, et al.: Cardiac Diseases Following Childhood Cancer Treatment: Cohort Study. Circulation 133 (1): 31-8, 2016. [PUBMED Abstract]
Cotton CA, Peterson S, Norkool PA, et al.: Early and late mortality after diagnosis of wilms tumor. J Clin Oncol 27 (8): 1304-9, 2009. [PUBMED Abstract]
Schellong G, Riepenhausen M, Bruch C, et al.: Late valvular and other cardiac diseases after different doses of mediastinal radiotherapy for Hodgkin disease in children and adolescents: report from the longitudinal GPOH follow-up project of the German-Austrian DAL-HD studies. Pediatr Blood Cancer 55 (6): 1145-52, 2010. [PUBMED Abstract]
Green DM, Kun LE, Matthay KK, et al.: Relevance of historical therapeutic approaches to the contemporary treatment of pediatric solid tumors. Pediatr Blood Cancer 60 (7): 1083-94, 2013. [PUBMED Abstract]
Hudson MM, Neglia JP, Woods WG, et al.: Lessons from the past: opportunities to improve childhood cancer survivor care through outcomes investigations of historical therapeutic approaches for pediatric hematological malignancies. Pediatr Blood Cancer 58 (3): 334-43, 2012. [PUBMED Abstract]
de Baat EC, van Dalen EC, Mulder RL, et al.: Primary cardioprotection with dexrazoxane in patients with childhood cancer who are expected to receive anthracyclines: recommendations from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Child Adolesc Health 6 (12): 885-894, 2022. [PUBMED Abstract]
Chow EJ, Antal Z, Constine LS, et al.: New Agents, Emerging Late Effects, and the Development of Precision Survivorship. J Clin Oncol 36 (21): 2231-2240, 2018. [PUBMED Abstract]
de Baat EC, Feijen EAM, Reulen RC, et al.: Risk Factors for Heart Failure Among Pan-European Childhood Cancer Survivors: A PanCareSurFup and ProCardio Cohort and Nested Case-Control Study. J Clin Oncol 41 (1): 96-106, 2023. [PUBMED Abstract]
Mulrooney DA, Hyun G, Ness KK, et al.: Major cardiac events for adult survivors of childhood cancer diagnosed between 1970 and 1999: report from the Childhood Cancer Survivor Study cohort. BMJ 368: l6794, 2020. [PUBMED Abstract]
Bates JE, Howell RM, Liu Q, et al.: Therapy-Related Cardiac Risk in Childhood Cancer Survivors: An Analysis of the Childhood Cancer Survivor Study. J Clin Oncol 37 (13): 1090-1101, 2019. [PUBMED Abstract]
Bates JE, Shrestha S, Liu Q, et al.: Cardiac Substructure Radiation Dose and Risk of Late Cardiac Disease in Survivors of Childhood Cancer: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 41 (22): 3826-3838, 2023. [PUBMED Abstract]
Chow EJ, Chen Y, Armstrong GT, et al.: Underdiagnosis and Undertreatment of Modifiable Cardiovascular Risk Factors Among Survivors of Childhood Cancer. J Am Heart Assoc 11 (12): e024735, 2022. [PUBMED Abstract]
Goldberg JF, Hyun G, Ness KK, et al.: Dyslipidemia and cardiovascular disease among childhood cancer survivors: a St. Jude Lifetime Cohort report. J Natl Cancer Inst 116 (3): 408-420, 2024. [PUBMED Abstract]
Kooijmans ECM, van der Pal HJH, Pluijm SMF, et al.: Hypertension in long-term childhood cancer survivors after treatment with potentially nephrotoxic therapy; DCCSS-LATER 2: Renal study. Eur J Cancer 172: 287-299, 2022. [PUBMED Abstract]
Bhakta N, Liu Q, Yeo F, et al.: Cumulative burden of cardiovascular morbidity in paediatric, adolescent, and young adult survivors of Hodgkin's lymphoma: an analysis from the St Jude Lifetime Cohort Study. Lancet Oncol 17 (9): 1325-34, 2016. [PUBMED Abstract]
Mulrooney DA, Soliman EZ, Ehrhardt MJ, et al.: Electrocardiographic abnormalities and mortality in aging survivors of childhood cancer: A report from the St Jude Lifetime Cohort Study. Am Heart J 189: 19-27, 2017. [PUBMED Abstract]
Feijen EAML, Font-Gonzalez A, Van der Pal HJH, et al.: Risk and Temporal Changes of Heart Failure Among 5-Year Childhood Cancer Survivors: a DCOG-LATER Study. J Am Heart Assoc 8 (1): e009122, 2019. [PUBMED Abstract]
Bates JE, Rancati T, Keshavarz H, et al.: Cardiac Disease in Childhood Cancer Survivors Treated With Radiation Therapy: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 522-532, 2024. [PUBMED Abstract]
van der Pal HJ, van Dalen EC, van Delden E, et al.: High risk of symptomatic cardiac events in childhood cancer survivors. J Clin Oncol 30 (13): 1429-37, 2012. [PUBMED Abstract]
Zhang S, Liu X, Bawa-Khalfe T, et al.: Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med 18 (11): 1639-42, 2012. [PUBMED Abstract]
Lyu YL, Kerrigan JE, Lin CP, et al.: Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res 67 (18): 8839-46, 2007. [PUBMED Abstract]
Hudson MM, Rai SN, Nunez C, et al.: Noninvasive evaluation of late anthracycline cardiac toxicity in childhood cancer survivors. J Clin Oncol 25 (24): 3635-43, 2007. [PUBMED Abstract]
van der Pal HJ, van Dalen EC, Hauptmann M, et al.: Cardiac function in 5-year survivors of childhood cancer: a long-term follow-up study. Arch Intern Med 170 (14): 1247-55, 2010. [PUBMED Abstract]
Ehrhardt MJ, Leerink JM, Mulder RL, et al.: Systematic review and updated recommendations for cardiomyopathy surveillance for survivors of childhood, adolescent, and young adult cancer from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol 24 (3): e108-e120, 2023. [PUBMED Abstract]
Chow EJ, Chen Y, Kremer LC, et al.: Individual prediction of heart failure among childhood cancer survivors. J Clin Oncol 33 (5): 394-402, 2015. [PUBMED Abstract]
Armenian SH, Sun CL, Vase T, et al.: Cardiovascular risk factors in hematopoietic cell transplantation survivors: role in development of subsequent cardiovascular disease. Blood 120 (23): 4505-12, 2012. [PUBMED Abstract]
Feijen EA, Leisenring WM, Stratton KL, et al.: Equivalence Ratio for Daunorubicin to Doxorubicin in Relation to Late Heart Failure in Survivors of Childhood Cancer. J Clin Oncol 33 (32): 3774-80, 2015. [PUBMED Abstract]
Feijen EAM, Leisenring WM, Stratton KL, et al.: Derivation of Anthracycline and Anthraquinone Equivalence Ratios to Doxorubicin for Late-Onset Cardiotoxicity. JAMA Oncol 5 (6): 864-871, 2019. [PUBMED Abstract]
van Dalen EC, Michiels EM, Caron HN, et al.: Different anthracycline derivates for reducing cardiotoxicity in cancer patients. Cochrane Database Syst Rev (5): CD005006, 2010. [PUBMED Abstract]
Krauss AC, Gao X, Li L, et al.: FDA Approval Summary: (Daunorubicin and Cytarabine) Liposome for Injection for the Treatment of Adults with High-Risk Acute Myeloid Leukemia. Clin Cancer Res 25 (9): 2685-2690, 2019. [PUBMED Abstract]
van Dalen EC, van der Pal HJ, Caron HN, et al.: Different dosage schedules for reducing cardiotoxicity in cancer patients receiving anthracycline chemotherapy. Cochrane Database Syst Rev (4): CD005008, 2009. [PUBMED Abstract]
Lipshultz SE, Giantris AL, Lipsitz SR, et al.: Doxorubicin administration by continuous infusion is not cardioprotective: the Dana-Farber 91-01 Acute Lymphoblastic Leukemia protocol. J Clin Oncol 20 (6): 1677-82, 2002. [PUBMED Abstract]
Hensley ML, Hagerty KL, Kewalramani T, et al.: American Society of Clinical Oncology 2008 clinical practice guideline update: use of chemotherapy and radiation therapy protectants. J Clin Oncol 27 (1): 127-45, 2009. [PUBMED Abstract]
van Dalen EC, Caron HN, Dickinson HO, et al.: Cardioprotective interventions for cancer patients receiving anthracyclines. Cochrane Database Syst Rev (6): CD003917, 2011. [PUBMED Abstract]
Wexler LH, Andrich MP, Venzon D, et al.: Randomized trial of the cardioprotective agent ICRF-187 in pediatric sarcoma patients treated with doxorubicin. J Clin Oncol 14 (2): 362-72, 1996. [PUBMED Abstract]
Lipshultz SE, Scully RE, Lipsitz SR, et al.: Assessment of dexrazoxane as a cardioprotectant in doxorubicin-treated children with high-risk acute lymphoblastic leukaemia: long-term follow-up of a prospective, randomised, multicentre trial. Lancet Oncol 11 (10): 950-61, 2010. [PUBMED Abstract]
Asselin BL, Devidas M, Chen L, et al.: Cardioprotection and Safety of Dexrazoxane in Patients Treated for Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia or Advanced-Stage Lymphoblastic Non-Hodgkin Lymphoma: A Report of the Children's Oncology Group Randomized Trial Pediatric Oncology Group 9404. J Clin Oncol 34 (8): 854-62, 2016. [PUBMED Abstract]
Shaikh F, Dupuis LL, Alexander S, et al.: Cardioprotection and Second Malignant Neoplasms Associated With Dexrazoxane in Children Receiving Anthracycline Chemotherapy: A Systematic Review and Meta-Analysis. J Natl Cancer Inst 108 (4): , 2016. [PUBMED Abstract]
Schwartz CL, Constine LS, Villaluna D, et al.: A risk-adapted, response-based approach using ABVE-PC for children and adolescents with intermediate- and high-risk Hodgkin lymphoma: the results of P9425. Blood 114 (10): 2051-9, 2009. [PUBMED Abstract]
Chow EJ, Aplenc R, Vrooman LM, et al.: Late health outcomes after dexrazoxane treatment: A report from the Children's Oncology Group. Cancer 128 (4): 788-796, 2022. [PUBMED Abstract]
Chow EJ, Aggarwal S, Doody DR, et al.: Dexrazoxane and Long-Term Heart Function in Survivors of Childhood Cancer. J Clin Oncol 41 (12): 2248-2257, 2023. [PUBMED Abstract]
Mansouri I, Allodji RS, Hill C, et al.: The role of irradiated heart and left ventricular volumes in heart failure occurrence after childhood cancer. Eur J Heart Fail 21 (4): 509-518, 2019. [PUBMED Abstract]
Shrestha S, Bates JE, Liu Q, et al.: Radiation therapy related cardiac disease risk in childhood cancer survivors: Updated dosimetry analysis from the Childhood Cancer Survivor Study. Radiother Oncol 163: 199-208, 2021. [PUBMED Abstract]
Lo AC, Liu A, Liu Q, et al.: Late Cardiac Toxic Effects Associated With Treatment Protocols for Hodgkin Lymphoma in Children. JAMA Netw Open 7 (1): e2351062, 2024. [PUBMED Abstract]
Passos J, Nzwalo H, Valente M, et al.: Microbleeds and cavernomas after radiotherapy for paediatric primary brain tumours. J Neurol Sci 372: 413-416, 2017. [PUBMED Abstract]
Bowers DC, Liu Y, Leisenring W, et al.: Late-occurring stroke among long-term survivors of childhood leukemia and brain tumors: a report from the Childhood Cancer Survivor Study. J Clin Oncol 24 (33): 5277-82, 2006. [PUBMED Abstract]
De Bruin ML, Dorresteijn LD, van't Veer MB, et al.: Increased risk of stroke and transient ischemic attack in 5-year survivors of Hodgkin lymphoma. J Natl Cancer Inst 101 (13): 928-37, 2009. [PUBMED Abstract]
van Dijk IW, van der Pal HJ, van Os RM, et al.: Risk of Symptomatic Stroke After Radiation Therapy for Childhood Cancer: A Long-Term Follow-Up Cohort Analysis. Int J Radiat Oncol Biol Phys 96 (3): 597-605, 2016. [PUBMED Abstract]
Reulen RC, Guha J, Bright CJ, et al.: Risk of cerebrovascular disease among 13 457 five-year survivors of childhood cancer: A population-based cohort study. Int J Cancer 148 (3): 572-583, 2021. [PUBMED Abstract]
Waxer JF, Wong K, Modiri A, et al.: Risk of Cerebrovascular Events Among Childhood and Adolescent Patients Receiving Cranial Radiation Therapy: A PENTEC Normal Tissue Outcomes Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 417-430, 2024. [PUBMED Abstract]
Mueller S, Sear K, Hills NK, et al.: Risk of first and recurrent stroke in childhood cancer survivors treated with cranial and cervical radiation therapy. Int J Radiat Oncol Biol Phys 86 (4): 643-8, 2013. [PUBMED Abstract]
Fullerton HJ, Stratton K, Mueller S, et al.: Recurrent stroke in childhood cancer survivors. Neurology 85 (12): 1056-64, 2015. [PUBMED Abstract]
Chow EJ, Chen Y, Hudson MM, et al.: Prediction of Ischemic Heart Disease and Stroke in Survivors of Childhood Cancer. J Clin Oncol 36 (1): 44-52, 2018. [PUBMED Abstract]
El-Fayech C, Haddy N, Allodji RS, et al.: Cerebrovascular Diseases in Childhood Cancer Survivors: Role of the Radiation Dose to Willis Circle Arteries. Int J Radiat Oncol Biol Phys 97 (2): 278-286, 2017. [PUBMED Abstract]
Bright CJ, Hawkins MM, Guha J, et al.: Risk of Cerebrovascular Events in 178 962 Five-Year Survivors of Cancer Diagnosed at 15 to 39 Years of Age: The TYACSS (Teenage and Young Adult Cancer Survivor Study). Circulation 135 (13): 1194-1210, 2017. [PUBMED Abstract]
Mueller S, Kline CN, Buerki RA, et al.: Stroke impact on mortality and psychologic morbidity within the Childhood Cancer Survivor Study. Cancer 126 (5): 1051-1059, 2020. [PUBMED Abstract]
Walker AJ, Grainge MJ, Card TR, et al.: Venous thromboembolism in children with cancer - a population-based cohort study. Thromb Res 133 (3): 340-4, 2014. [PUBMED Abstract]
Madenci AL, Weil BR, Liu Q, et al.: Long-Term Risk of Venous Thromboembolism in Survivors of Childhood Cancer: A Report From the Childhood Cancer Survivor Study. J Clin Oncol : JCO2018784595, 2018. [PUBMED Abstract]
Mueller S, Fullerton HJ, Stratton K, et al.: Radiation, atherosclerotic risk factors, and stroke risk in survivors of pediatric cancer: a report from the Childhood Cancer Survivor Study. Int J Radiat Oncol Biol Phys 86 (4): 649-55, 2013. [PUBMED Abstract]
Chao C, Xu L, Bhatia S, et al.: Cardiovascular Disease Risk Profiles in Survivors of Adolescent and Young Adult (AYA) Cancer: The Kaiser Permanente AYA Cancer Survivors Study. J Clin Oncol 34 (14): 1626-33, 2016. [PUBMED Abstract]
Winther JF, Bhatia S, Cederkvist L, et al.: Risk of cardiovascular disease among Nordic childhood cancer survivors with diabetes mellitus: A report from adult life after childhood cancer in Scandinavia. Cancer 124 (22): 4393-4400, 2018. [PUBMED Abstract]
van Dalen EC, Mulder RL, Suh E, et al.: Coronary artery disease surveillance among childhood, adolescent and young adult cancer survivors: A systematic review and recommendations from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Eur J Cancer 156: 127-137, 2021. [PUBMED Abstract]
Lipshultz SE, Lipsitz SR, Kutok JL, et al.: Impact of hemochromatosis gene mutations on cardiac status in doxorubicin-treated survivors of childhood high-risk leukemia. Cancer 119 (19): 3555-62, 2013. [PUBMED Abstract]
Visscher H, Ross CJ, Rassekh SR, et al.: Validation of variants in SLC28A3 and UGT1A6 as genetic markers predictive of anthracycline-induced cardiotoxicity in children. Pediatr Blood Cancer 60 (8): 1375-81, 2013. [PUBMED Abstract]
Wang X, Liu W, Sun CL, et al.: Hyaluronan synthase 3 variant and anthracycline-related cardiomyopathy: a report from the children's oncology group. J Clin Oncol 32 (7): 647-53, 2014. [PUBMED Abstract]
Wang X, Sun CL, Quiñones-Lombraña A, et al.: CELF4 Variant and Anthracycline-Related Cardiomyopathy: A Children's Oncology Group Genome-Wide Association Study. J Clin Oncol 34 (8): 863-70, 2016. [PUBMED Abstract]
Aminkeng F, Bhavsar AP, Visscher H, et al.: A coding variant in RARG confers susceptibility to anthracycline-induced cardiotoxicity in childhood cancer. Nat Genet 47 (9): 1079-84, 2015. [PUBMED Abstract]
Visscher H, Rassekh SR, Sandor GS, et al.: Genetic variants in SLC22A17 and SLC22A7 are associated with anthracycline-induced cardiotoxicity in children. Pharmacogenomics 16 (10): 1065-76, 2015. [PUBMED Abstract]
Singh P, Wang X, Hageman L, et al.: Association of GSTM1 null variant with anthracycline-related cardiomyopathy after childhood cancer-A Children's Oncology Group ALTE03N1 report. Cancer 126 (17): 4051-4058, 2020. [PUBMED Abstract]
Sapkota Y, Ehrhardt MJ, Qin N, et al.: A Novel Locus on 6p21.2 for Cancer Treatment-Induced Cardiac Dysfunction Among Childhood Cancer Survivors. J Natl Cancer Inst 114 (8): 1109-1116, 2022. [PUBMED Abstract]
Davies SM: Getting to the heart of the matter. J Clin Oncol 30 (13): 1399-400, 2012. [PUBMED Abstract]
Hines MR, Mulrooney DA, Hudson MM, et al.: Pregnancy-associated cardiomyopathy in survivors of childhood cancer. J Cancer Surviv 10 (1): 113-21, 2016. [PUBMED Abstract]
Chait-Rubinek L, Mariani JA, Goroncy N, et al.: A Retrospective Evaluation of Risk of Peripartum Cardiac Dysfunction in Survivors of Childhood, Adolescent and Young Adult Malignancies. Cancers (Basel) 11 (8): , 2019. [PUBMED Abstract]
Liu S, Aghel N, Belford L, et al.: Cardiac Outcomes in Pregnant Women With Treated Cancer. J Am Coll Cardiol 72 (17): 2087-2089, 2018. [PUBMED Abstract]
Bottinor W, Im C, Doody DR, et al.: Mortality After Major Cardiovascular Events in Survivors of Childhood Cancer. J Am Coll Cardiol 83 (8): 827-838, 2024. [PUBMED Abstract]
Dietz AC, Seidel K, Leisenring WM, et al.: Solid organ transplantation after treatment for childhood cancer: a retrospective cohort analysis from the Childhood Cancer Survivor Study. Lancet Oncol 20 (10): 1420-1431, 2019. [PUBMED Abstract]
Brouwer CA, Postma A, Hooimeijer HL, et al.: Endothelial damage in long-term survivors of childhood cancer. J Clin Oncol 31 (31): 3906-13, 2013. [PUBMED Abstract]
Maraldo MV, Jørgensen M, Brodin NP, et al.: The impact of involved node, involved field and mantle field radiotherapy on estimated radiation doses and risk of late effects for pediatric patients with Hodgkin lymphoma. Pediatr Blood Cancer 61 (4): 717-22, 2014. [PUBMED Abstract]
Tukenova M, Guibout C, Oberlin O, et al.: Role of cancer treatment in long-term overall and cardiovascular mortality after childhood cancer. J Clin Oncol 28 (8): 1308-15, 2010. [PUBMED Abstract]
Armstrong GT, Kawashima T, Leisenring W, et al.: Aging and risk of severe, disabling, life-threatening, and fatal events in the childhood cancer survivor study. J Clin Oncol 32 (12): 1218-27, 2014. [PUBMED Abstract]
Chen AB, Punglia RS, Kuntz KM, et al.: Cost effectiveness and screening interval of lipid screening in Hodgkin's lymphoma survivors. J Clin Oncol 27 (32): 5383-9, 2009. [PUBMED Abstract]
Wong FL, Bhatia S, Landier W, et al.: Cost-effectiveness of the children's oncology group long-term follow-up screening guidelines for childhood cancer survivors at risk for treatment-related heart failure. Ann Intern Med 160 (10): 672-83, 2014. [PUBMED Abstract]
Yeh JM, Nohria A, Diller L: Routine echocardiography screening for asymptomatic left ventricular dysfunction in childhood cancer survivors: a model-based estimation of the clinical and economic effects. Ann Intern Med 160 (10): 661-71, 2014. [PUBMED Abstract]
Landier W, Armenian SH, Lee J, et al.: Yield of screening for long-term complications using the children's oncology group long-term follow-up guidelines. J Clin Oncol 30 (35): 4401-8, 2012. [PUBMED Abstract]
Ramjaun A, AlDuhaiby E, Ahmed S, et al.: Echocardiographic Detection of Cardiac Dysfunction in Childhood Cancer Survivors: How Long Is Screening Required? Pediatr Blood Cancer 62 (12): 2197-203, 2015. [PUBMED Abstract]
Spewak MB, Williamson RS, Mertens AC, et al.: Yield of screening echocardiograms during pediatric follow-up in survivors treated with anthracyclines and cardiotoxic radiation. Pediatr Blood Cancer 64 (6): , 2017. [PUBMED Abstract]
Chen Y, Chow EJ, Oeffinger KC, et al.: Traditional Cardiovascular Risk Factors and Individual Prediction of Cardiovascular Events in Childhood Cancer Survivors. J Natl Cancer Inst 112 (3): 256-265, 2020. [PUBMED Abstract]
Scott JM, Li N, Liu Q, et al.: Association of Exercise With Mortality in Adult Survivors of Childhood Cancer. JAMA Oncol 4 (10): 1352-1358, 2018. [PUBMED Abstract]
Schindera C, Zürcher SJ, Jung R, et al.: Physical fitness and modifiable cardiovascular disease risk factors in survivors of childhood cancer: A report from the SURfit study. Cancer 127 (10): 1690-1698, 2021. [PUBMED Abstract]
Rueegg CS, Zürcher SJ, Schindera C, et al.: Effect of a 1-year physical activity intervention on cardiovascular health in long-term childhood cancer survivors-a randomised controlled trial (SURfit). Br J Cancer 129 (8): 1284-1297, 2023. [PUBMED Abstract]
Efectos tardíos en el sistema nervioso central
Neurocognitivos
Los efectos tardíos neurocognitivos se observan con mayor frecuencia después del tratamiento de neoplasias malignas que exigen tratamientos dirigidos al sistema nervioso central (SNC), entre los que se incluyen los siguientes:
Radioterapia craneal.
Quimioterapia sistémica con dosis altas de metotrexato o citarabina.
Los niños con tumores del SNC o leucemia linfoblástica aguda (LLA) son quienes, con más probabilidad, resulten afectados. Los factores de riesgo de los efectos secundarios neurocognitivos son los siguientes:[3-7]
Sexo femenino.
Edad más temprana en el momento del tratamiento.
Localización del tumor.
Tratamiento con radioterapia craneal y fármacos quimioterapéuticos (sistémicos o intratecales).
Dosis más alta de radiación craneal.
Tiempo desde el tratamiento.
Los fenotipos cognitivos observados en los niños sobrevivientes de LLA y tumores del SNC a veces difieren de los trastornos del desarrollo tradicionales. Por ejemplo, el fenotipo de los problemas de atención en los sobrevivientes de LLA y tumores de encéfalo difiere del trastorno por déficit de atención o hiperactividad (TDAH) del desarrollo debido a que pocos sobrevivientes muestran hiperactividad o impulsividad importantes y, en cambio, tienen dificultades relacionadas con la velocidad de procesamiento y el funcionamiento ejecutivo.[8,9]
El Pediatric Normal Tissue Effects in the Clinic (PENTEC) realizó una revisión exhaustiva con el fin de crear modelos que facilitan la identificación de las restricciones de dosis para las morbilidades del SNC relacionadas con la radiación.[10]
Los modelos indican un riesgo del 5 % de presentar un cociente intelectual (CI) posterior inferior a 85 cuando sucede lo siguiente:
El 10 % del encéfalo se irradia con 35,7 Gy.
El 20 % del encéfalo se irradia con 29,1 Gy.
El 50 % del encéfalo se irradia con 22,2 Gy.
El 100 % del encéfalo se irradia con 18,1 Gy.
Nota: todos a 2 Gy/fracción y sin metotrexato.
La exposición al metotrexato aumentó el riesgo de CI inferior a 85, de forma similar a como sucede con una dosis de radiación cerebral uniforme generalizada de 5,9 Gy. No obstante, la falta de datos sobre las dosis totales de metotrexato, tanto intravenosas como intratecales, fue una limitación de este análisis.
El modelo para predecir el CI esperado, que también incluía el efecto de la dosis, la edad y el metotrexato, indicó que cada uno de estos factores tiene un efecto independiente, pero tal vez acumulativo, sobre el CI.
Además de los efectos directos de los tratamientos neurotóxicos, como la radiación craneal, los investigadores del Childhood Cancer Survivor Study (CCSS) observaron que las afecciones crónicas que resultan de las exposiciones a tratamientos que no son neurotóxicos (por ejemplo, radiación torácica) pueden afectar de modo adverso el funcionamiento neurocognitivo, debido a disfunción cardiopulmonar y endocrina.[11] Además, algunas secuelas de la terapia neurotóxica (por ejemplo, hipoacusia grave) se han relacionado con déficits neurocognitivos independientes del tratamiento neurotóxico recibido.[12]
En una investigación relacionada del CCSS se evaluaron asociaciones longitudinales entre la actividad física y los problemas neurocognitivos en adultos sobrevivientes de cáncer infantil.[13]
Los sobrevivientes eran menos propensos a notificar actividad física constante (28,1 vs. 33,6 %) en comparación con sus hermanos o hermanas.
Los sobrevivientes que notificaron más actividad física constante presentaron menos problemas neurocognitivos y mayores mejoras de problemas neurocognitivos años después del tratamiento.
El índice de masa corporal (IMC) y las afecciones crónicas graves afectaron, en cierta medida, la relación entre la actividad física y las capacidades neurocognitivas, pero este efecto fue pequeño.
En una revisión sistemática y un metanálisis posteriores se compararon los efectos de las intervenciones de actividad o ejercicio físico sobre la función cognitiva en personas diagnosticadas de cáncer (de 0 a 19 años) con los de los controles. Se encontraron 22 estudios únicos (16 ensayos controlados aleatorizados) con datos de 12 767 personas.[14] La mediana de edad al comienzo del estudio fue de 12 años (intervalo intercuartil [RIC], 11–14 años), la mediana de edad desde la finalización del tratamiento del cáncer fue de 2,5 años (RIC, 1,1–3,0 años) y la mediana del período de intervención fue de 12 semanas (RIC, 10–24 semanas).
En comparación con los controles, hubo evidencia de calidad moderada de que la actividad y el ejercicio físico mejoraron las medidas de rendimiento cognitivo y de la función cognitiva en los sobrevivientes de cáncer según la notificación de los pacientes.
Es posible que los sobrevivientes de cáncer infantil tengan riesgo de presentar deterioro cognitivo a lo largo de la vida (aunque no se manifieste en los primeros 10 años después del tratamiento). En un estudio de 2375 sobrevivientes adultos de LLA infantil, linfoma de Hodgkin o tumores del SNC (edad media en el momento de la evaluación, 31,8 años) y los hermanos del grupo de control, el deterioro de la memoria de nueva aparición fue más frecuente en los sobrevivientes, décadas después del diagnóstico y el tratamiento del cáncer.[15]
En comparación con los hermanos, una mayor proporción de sobrevivientes sin deterioro de la memoria al inicio del estudio presentaron deterioro de la memoria de nueva aparición durante el seguimiento, de la siguiente manera:
Porcentaje de hermanos, 7,8 %.
Sobrevivientes de LLA tratados con quimioterapia sola, 14,0 %.
Sobrevivientes de LLA tratados con radioterapia craneal, 25,8 %.
Sobrevivientes de tumores del SNC, 34,7 %.
Sobrevivientes de linfoma de Hodgkin, 16,6 %.
La factores de riesgo relacionados con trastorno de memoria de nueva aparición incluso radiación craneal en sobrevivientes de tumores del SNC (riesgo relativo [RR], 1,97) y dosis de 8000 mg/m2 o más de quimioterapia con alquilantes en sobrevivientes de LLA tratados sin radioterapia craneal (RR, 2,80).
Las condiciones neurológicas mediaron el efecto de la radioterapia craneal en el deterioro de la memoria de nueva aparición en sobrevivientes de tumores del SNC.
El consumo de tabaco, la obesidad, el nivel educativo bajo y la actividad física escasa se relacionaron con un mayor riesgo de aparición de un nuevo deterioro de la memoria.
Desenlaces neurocognitivos en sobrevivientes de tumores de encéfalo
Los efectos cognitivos a largo plazo causados por la enfermedad y los tratamientos administrados son efectos de morbilidad bien establecidos en sobrevivientes de tumores de encéfalo durante la niñez y la adolescencia. En este grupo, los factores de riesgo para los efectos adversos neurocognitivos son los siguientes:
Radioterapia craneal.
Este tipo de radioterapia se ha relacionado con el riesgo más alto de morbilidad cognitiva a largo plazo, en particular, en los niños más pequeños.[3,4]
Hay una relación establecida entre la dosis y la respuesta, conforme a la cual los pacientes que reciben las dosis más altas de radiación craneal sistemáticamente obtienen resultados más adversos en las mediciones intelectuales.[3,4,16]
Se ha observado que la dosis de radiación dirigida a regiones específicas del encéfalo como los lóbulos temporales y el hipocampo, afecta de manera significativa las puntuaciones longitudinales del CI y las puntuaciones de los logros académicos en los niños tratados con irradiación craneoespinal por un meduloblastoma.[17]
El efecto negativo del tratamiento con radiación se caracterizó por cambios en las puntuaciones de CI, que cayeron alrededor de entre 2 a 5 años después del diagnóstico.[24-26]
La disminución continúa 5 a 10 años después; sin embargo, se tiene menos información sobre la posible estabilización o la disminución adicional de las puntuaciones de CI varias décadas después del diagnóstico.[24,26]
La disminución de las puntuaciones de CI con el paso del tiempo suele reflejar más la incapacidad del niño de adquirir nuevas destrezas o información a una velocidad similar a la de sus pares, que una pérdida progresiva de destrezas y conocimientos.[27]
Los niños afectados tal vez también presenten alteraciones en otras áreas cognitivas, incluso en el ámbito académico (lectura y matemáticas) y problemas de atención, velocidad de procesamiento, memoria, y destrezas motoras, visuales o perceptuales.[28]
Los cambios en el funcionamiento cognitivo se explican, en parte, por la reducción (inducida por la radiación) del volumen de sustancia blanca con apariencia normal o la integridad de las vías de la sustancia blanca evaluada mediante imágenes por resonancia magnética (IRM).[29,30]
La reducción de la integridad de la sustancia blanca se vinculó de forma directa con una menor velocidad de procesamiento cognitivo en los sobrevivientes de tumores de encéfalo,[31] mientras que un mayor volumen de sustancia blanca se relacionó con una mejor memoria operativa; sobre todo en las mujeres.[30]
Los datos de los protocolos actuales muestran que el uso de dosis más bajas de radiación craneal, radiación con haz de protones y volúmenes de tratamiento más precisos reducen la gravedad de los efectos neurocognitivos del tratamiento.[3,19]
Evidencia (factores de predicción de deterioro cognitivo en sobrevivientes de tumores del SNC):
Los estudios de cohortes longitudinales han permitido entender la evolución y los factores pronósticos del deterioro cognitivo en sobrevivientes de tumores del SNC.
En un ensayo prospectivo longitudinal multisitio se evaluaron predictores de desempeño cognitivo en 139 lactantes con tumores de encéfalo que recibieron quimioterapia con radioterapia focal de protones o fotones, o sin esta.[32]
El CI, la memoria operativa notificada por un progenitor y la función adaptativa notificada por un progenitor fueron más bajas que las expectativas al inicio; los factores predictivos fueron la edad más temprana y un nivel socioeconómico más bajo.
El CI permaneció estable en el tiempo, mientras que aumentaron la atención notificada por un progenitor y la alteración de la función ejecutiva.
Los desenlaces cognitivos no difirieron según la exposición al tratamiento (quimioterapia sola vs. quimioterapia con radioterapia).
Los cambios en el funcionamiento cognitivo se relacionaron con una ubicación supratentorial del tumor y una derivación del líquido cefalorraquídeo.
En el St. Jude Children’s Research Hospital (SJCRH), se estudió a 78 pacientes menores de 20 años (media de 9,7 años) con diagnóstico de glioma de grado bajo.[33]
Se observó deterioro cognitivo después de la administración de 54 Gy de radioterapia craneal conformada (consultar la Figura 5).
La edad en el momento de la radiación craneal fue más importante que la dosis para pronosticar un deterioro cognitivo y se estimó que los niños menores de 5 años tendrían el mayor deterioro cognitivo.
En un estudio de 51 niños con gliomas de grado bajo y tumores glioneurales de grado bajo diagnosticados durante el primer año de vida, se notificaron los siguientes resultados:[34]
Media de puntuación de CI de 75,5; el 75 % de los niños tenían puntuaciones de CI inferiores a 85.
Los factores de predicción de un CI bajo fueron la localización supratentorial del tumor primario y el tratamiento con más regímenes de quimioterapia, pero no el uso de radiación.
La capacidad del niño de completar tareas apropiadas para la edad se afectó de la misma manera que las puntuaciones de CI.
En un estudio de 126 sobrevivientes de meduloblastoma tratados con 23,4 Gy o 36 a 39,6 Gy de radiación craneoespinal (con una dosis conformada de refuerzo de 55,8 Gy dirigidos al lecho del tumor primario), se evaluó la velocidad de procesamiento, la atención y el funcionamiento de la memoria.[35]
Las puntuaciones de velocidad de procesamiento disminuyeron de manera significativa con el tiempo, pero se observó menos deterioro funcional en la atención y la memoria. Las dosis de radiación más altas y la edad menor en el momento del diagnóstico pronosticaron una velocidad de procesamiento más lenta a lo largo del tiempo.
Los estudios de memoria operativa y logros académicos de pacientes inscritos en el mismo ensayo de meduloblastoma (SJMB03 [NCT00085202] del SJCRH), indicaron que el rendimiento se ubicó, en gran parte, en el intervalo de edad esperado hasta 5 años después del diagnóstico,[36,37] pero en ambos estudios, un síndrome de fosa posterior, una dosis más alta de radiación craneal y una edad menor en el momento del diagnóstico pronosticaron un desempeño más precario con el paso del tiempo.
La hipoacusia grave se relacionó con un deterioro intelectual y académico con el paso del tiempo.[37]
En un estudio prospectivo de 178 niños con meduloblastoma, 60 (34 %) presentaron síndrome de la fosa posterior. De esos pacientes, 40 (23 %) presentaron mutismo completo y 20 (11 %) disminución del habla.[38]
Todos los niños con síndrome de fosa posterior presentaron ataxia grave, y el 42,5 % de los pacientes con síndrome de fosa posterior y mutismo completo tenían trastornos del movimiento.
Los factores de riesgo independientes para el síndrome de la fosa posterior incluyeron una edad más temprana y la cirugía en un centro quirúrgico con bajo volumen de pacientes. El riesgo fue menor entre los niños con tumores con activación de erizo sónico (sonic hedgehog [SHH]).
Los niños con síndrome de fosa posterior y mutismo completo recuperaron el habla y la capacidad de marcha al cabo de una mediana de 2,3 y 0,7 meses, respectivamente. Los niños con síndrome de fosa posterior y habla disminuida recuperaron el habla y la capacidad de marcha al cabo de una mediana de 2,1 y 1,5 meses, respectivamente. Sin embargo, después de 12 meses de seguimiento, 12 de 27 niños (44,4 %) con síndrome de fosa posterior y mutismo completo no caminaban al cumplir 1 año de edad.
La presencia de un trastorno del movimiento o un puntaje elevado de ataxia se relacionó con un retraso en la recuperación del habla, mientras que la edad avanzada y un puntaje elevado de ataxia se asociaron con un retraso en la recuperación de la marcha.
Los síntomas mejoraron en todos los niños, pero ningún niño con síndrome de la fosa posterior tuvo una exploración neurológica normal al cabo de una mediana de 23 meses tras la cirugía.
En un estudio prospectivo se compararon 36 pacientes de meduloblastoma infantil que presentaron síndrome de la fosa posterior con 36 pacientes de meduloblastoma que no presentaron este síndrome, pero que estuvieron emparejados según el tratamiento y la edad en el momento del diagnóstico.[38,39]
El grupo de síndrome de la fosa posterior presentó una media más baja para las puntuaciones de capacidad intelectual, velocidad de procesamiento, memoria operativa y relaciones espaciales, en comparación con el grupo sin síndrome de la fosa posterior después de 1, 3 y 5 años del diagnóstico.
El grupo con síndrome de la fosa posterior exhibió poca recuperación con el tiempo y empeoramiento adicional en algunos dominios (atención y memoria operativa), en comparación con el grupo sin síndrome de la fosa posterior.
Un grupo de investigadores canadienses evaluó el efecto de la radiación (volumen de dosis y refuerzo) y las complicaciones neurológicas en modelos de funcionamiento intelectual en una cohorte de 113 sobrevivientes de meduloblastoma (media de edad en el momento del diagnóstico, 7,5 años; media de tiempo desde el diagnóstico hasta la última evaluación, 6 años).[4]
Los sobrevivientes tratados con dosis reducidas de radioterapia craneoespinal y refuerzo al lecho del tumor exhibieron un funcionamiento intelectual estable.
Las complicaciones neurológicas, como la hidrocefalia que requiere derivación del líquido cefalorraquídeo y produce mutismo, y el tratamiento con dosis más altas y volúmenes de refuerzo más grandes produjeron una disminución intelectual con una evolución característica.
En los estudios se están empezando a examinar los desenlaces cognitivos en subtipos de tumores de encéfalo con características histológicas distintivas.
En los datos de una muestra de 121 pacientes de meduloblastoma se observó una variación en los desenlaces cognitivos de 4 subgrupos moleculares distintos y diferencias en los perfiles de cambio con el paso del tiempo.[40]
Se requiere más investigación para establecer si los desenlaces neurocognitivos varían entre los subtipos biológicos característicos de los tumores de encéfalo infantiles.
El nivel socioeconómico predice desenlaces cognitivos a largo plazo en sobrevivientes de tumores de encéfalo. En un estudio prospectivo longitudinal de fase II de 248 niños tratados con radioterapia conformada (54–59,4 Gy) por ependimoma, glioma de grado bajo o craneofaringioma, los pacientes se vigilaron de manera seriada con evaluaciones cognitivas durante 10 años.[23]
Al inicio, antes de la radioterapia, se observaron asociaciones significativas entre el nivel socioeconómico y los puntajes de CI, lectura y matemáticas, así como la atención sostenida y la función adaptativa. Un nivel socioeconómico más alto se asoció con mejor desempeño (P < 0,005).
El nivel socioeconómico predijo un cambio de los puntajes de CI, lectura y matemáticas que se observó con el tiempo. Un nivel socioeconómico más alto se asoció con menos deterioro (P < 0,001).
Estos resultados indican que el nivel socioeconómico más alto es un factor de protección de efectos tardíos cognitivos.
Evidencia (factores de predicción de deterioro cognitivo en sobrevivientes a largo plazo de tumores del SNC):
Aunque se presume que los efectos neurocognitivos adversos observados entre 5 y 10 años después del tratamiento son generalizados y quizás empeoren con el tiempo, se dispone de escasos datos empíricos sobre el funcionamiento neurocognitivo en sobrevivientes a muy largo plazo de tumores del SNC.
En un estudio longitudinal se evaluó la relación entre la dosis dirigida al hipocampo y el deterioro en la memoria a corto plazo en 80 niños y adolescentes con gliomas de grado bajo (mediana de edad, 9,5 años en el momento del tratamiento) que recibieron 54 Gy de radioterapia.[41]
Al cabo de una mediana de seguimiento neurocognitivo de 9,8 años, una dosis dirigida al hipocampo más alta (volumen recibido de 40 Gy) se relacionó con una reducción más profunda en el recuerdo diferido.
De los adultos sobrevivientes que participaron en el CCSS, los sobrevivientes de tumores del SNC (n = 802) notificaron muchos más problemas de atención o velocidad de procesamiento, memoria, control emocional y organización que los sobrevivientes de otras neoplasias malignas que no estaban ubicadas en el SNC (n = 5937) y los hermanos del grupo de control (n = 382).[42]
En otro estudio del CCSS donde se evaluaron los patrones de mortalidad y morbilidad en 2821 adultos sobrevivientes de tumores del SNC se notificó deterioro en las mediciones de atención o velocidad de procesamiento (42,9–73,3 %) y de memoria (14,3–37,4 %); se observaron diferencias según el diagnóstico y la dosis de radiación craneal.[43]
En un análisis del estudio de cohorte St. Jude Life (SJLIFE) con 224 adultos sobrevivientes de tumores de encéfalo en la niñez, se observó que del 20 % al 30 % de los sobrevivientes exhibieron deterioro neurocognitivo grave (definido como un mínimo de 2 desviaciones estándar por debajo de la media de referencia) en las evaluaciones clínicas de inteligencia, memoria y funcionamiento ejecutivo (por ejemplo, planificación, organización y flexibilidad).[3]
Entre los adultos de la población general, la tasa de deterioro esperada en este umbral es del 2 %.
Los sobrevivientes que recibieron irradiación craneal a todo el encéfalo tuvieron entre 1,5 y 3 veces más probabilidades de deterioro neurocognitivo grave que los sobrevivientes que no la recibieron.
La hidrocefalia con colocación de una derivación y las convulsiones también se relacionaron con un aumento del riesgo de deterioro.
En el CCSS, los investigadores compararon los desenlaces a largo plazo de aspectos neuropsicológicos y de nivel socioeconómico de 181 adultos sobrevivientes de gliomas infantiles de grado bajo con los desenlaces de un grupo de comparación compuesto por hermanos emparejados por edad y sexo.[44]
Los sobrevivientes tratados con cirugía y radioterapia (mediana de edad en el momento del diagnóstico de 7 años; mediana de edad en el momento de la evaluación de 41 años) tuvieron una puntuación más baja en el CI calculado que los sobrevivientes tratados con cirugía sola, que tuvieron una puntuación más baja que los hermanos (cirugía y radioterapia, 93,9; cirugía sola, 101,2; hermanos, 108,5; todos los valores de P < 0,0001).
La edad más temprana en el momento del diagnóstico predijo puntuaciones bajas en todos los desenlaces neuropsicológicos, excepto la velocidad de atención o procesamiento.
Los sobrevivientes tratados con cirugía y radioterapia tuvieron puntuaciones de empleo, ingreso y educación más de 2 veces por encima de las puntuaciones de los sobrevivientes tratados con cirugía sola.
En una revisión retrospectiva de 528 sobrevivientes de tumores de encéfalo diagnosticados entre 2000 y 2015, la prevalencia de un diagnóstico de TDAH fue del 13,1 %.[45]
De los sobrevivientes, el 12,1 % recibió medicación para el TDAH y el 19,9 % de los sobrevivientes tuvieron síntomas de TDAH sin un diagnóstico clínico.
El diagnóstico de TDAH se relacionó con una edad más joven en el momento del diagnóstico del tumor y una ubicación supratentorial del mismo, pero no con el sexo, el tipo del tumor ni el tipo de tratamiento.
Es posible que las consecuencias neurocognitivas de la enfermedad en el SNC y su tratamiento tengan un efecto considerable en los desenlaces funcionales de los sobrevivientes de tumores de encéfalo.
En la niñez y la adolescencia, las alteraciones neurocognitivas se han relacionado con un ajuste social deficiente, que incluye problemas para relacionarse con los pares, aislamiento social y disminución de las habilidades sociales.[46,47]
Los sobrevivientes de tumores del SNC tienen más probabilidades de necesitar servicios de educación especial que los sobrevivientes de otras neoplasias malignas.[48]
Los adultos sobrevivientes de tumores del SNC tienen menos probabilidades de vivir en forma independiente, casarse y graduarse de la universidad que los sobrevivientes de otras neoplasias malignas y los hermanos.[49]
Desenlaces cognitivos después de la radioterapia de protones
Están surgiendo datos acerca de los desenlaces cognitivos tras la radiación de protones dirigida al SNC.[50-52] Sin embargo, estos estudios se han visto limitados por el análisis retrospectivo de desenlaces cognitivos en cohortes relativamente pequeñas de pacientes con tumores de encéfalo infantiles heterogéneos desde el punto de vista clínico y por el uso de pacientes con antecedentes de tratamiento con fotones o de poblaciones estándar como grupos de comparación.
En los estudios que describen sobre todo los cambios en el CI durante el seguimiento temprano (<5 años desde la radiación), los resultados demuestran una ausencia de diferencia en las pendientes de cambio del CI entre los pacientes tratados con fotones y protones,[50] además de disminuciones significativas en la velocidad de procesamiento cognitivo en los pacientes tratados con radiación de protones.[51]
En un estudio se comparó la evolución intelectual entre pacientes pediátricos con meduloblastoma que recibieron radioterapia de protones y fotones (4,3 años de media de seguimiento después de una mediana de dosis de irradiación craneoespinal de 23,4 Gy). Cabe destacar que la dosis de refuerzo y los márgenes fueron significativamente diferentes entre los 2 grupos.[52]
Los niños que recibieron radioterapia de protones presentaron desenlaces superiores a largo plazo en el CI general, el razonamiento perceptivo y la memoria operativa en comparación con los niños que recibieron radioterapia con fotones.
El grupo de radioterapia con fotones mostró una disminución significativa en el CI general, la memoria operativa y la velocidad de procesamiento.
El grupo de radioterapia de protones mostró puntuaciones estables a lo largo del tiempo en todos los dominios, excepto en la velocidad de procesamiento.
Teniendo en cuenta el tiempo de seguimiento relativamente corto desde la radiación, el seguimiento longitudinal es importante para determinar si la radiación de protones ofrece un beneficio clínico apreciable en la protección del funcionamiento cognitivo en comparación con la radiación de fotones. Además, volúmenes de radiación de fotones dirigidos con más precisión quizás reduzcan las posibles diferencias.
Desenlaces neurocognitivos en sobrevivientes de leucemia linfoblástica aguda
A fin de reducir al máximo el riesgo de secuelas cognitivas tardías, para el tratamiento contemporáneo de la leucemia linfoblástica aguda (LLA) se utiliza un abordaje de riesgo estratificado que reserva la irradiación craneal para los niños que se consideran con riesgo alto de recaída en el SNC.
Leucemia linfoblástica aguda y radiación craneal
En los sobrevivientes de LLA, la radiación craneal quizás produzca secuelas neurológicas clínicas y radiográficas tardías como las siguientes:
Leucoencefalopatía clínica. La leucoencefalopatía clínica, caracterizada por encefalopatía fluctuante, síntomas similares a los del accidente cerebrovascular, espasticidad, ataxia, disartria, disfagia, hemiparesia y convulsiones, es poco frecuente después del tratamiento contemporáneo de la LLA. Por el contrario, las neuroimágenes suelen mostrar anomalías en la sustancia blanca en sobrevivientes tratados con irradiación craneal o dosis altas de metotrexato.
Se notificó leucoencefalopatía radiográfica en hasta el 80 % de los niños que recibieron algún régimen de tratamiento.
Se informó que las dosis más altas y los ciclos más numerosos de metotrexato intravenoso aumentan el riesgo de leucoencefalopatía.[53]
En muchos pacientes, las anomalías en la sustancia blanca son transitorias y su prevalencia, grado e intensidad disminuyen cuando pasa más tiempo después de completar el tratamiento.[53]
La leucoencefalopatía produce volúmenes reducidos de sustancia blanca que se correlacionaron con deficiencias cognitivas.[53]
Aunque estas anomalías son leves en los pacientes irradiados (disminución general del CI de casi 10 puntos), quienes recibieron dosis más altas a una edad temprana tal vez presenten dificultades de aprendizaje importantes.[54,55]
Deficiencias neurocognitivas. Hay informes sobre deficiencias del funcionamiento neurocognitivo, en aspectos como la integración visomotriz, la velocidad de procesamiento, la atención y la memoria a corto plazo, en niños tratados con 18 a 24 Gy.[54,56,57]
Las mujeres y los niños tratados en edades más tempranas son más vulnerables a los efectos adversos de la radiación craneal en el encéfalo en desarrollo.[5]
El deterioro del funcionamiento intelectual parece ser progresivo, con mayor disminución del funcionamiento cognitivo a medida que aumenta el tiempo transcurrido desde la administración de la radioterapia.[5,6]
En unos pocos estudios se indica que los sobrevivientes a largo plazo de LLA infantil tratados con irradiación craneal tienen riesgo de deterioro cognitivo progresivo compatible con un deterioro cognitivo leve de inicio temprano; este riesgo es más elevado en los pacientes tratados con dosis de 24 Gy de radiación craneal.[58,59]
Leucemia linfoblástica aguda y tratamiento del sistema nervioso central con quimioterapia sola
Debido a su penetrancia en el sistema nervioso central (SNC), se ha utilizado el metotrexato sistémico en una variedad de regímenes profilácticos de dosis bajas y altas para tratar la leucemia en el SNC.
El metotrexato sistémico en dosis altas, combinado con radioterapia o sin esta, puede conducir a una leucoencefalopatía infrecuente pero bien descrita, la cual se relacionó con deterioro cognitivo.[53]
Cuando se comparan en forma directa los desenlaces neurocognitivos posteriores a los regímenes de radioterapia y quimioterapia sola, se aprecia un mejor desenlace para los pacientes tratados con quimioterapia sola, aunque en algunos estudios no se observa ninguna diferencia significativa.[60,61]
En un análisis longitudinal de 210 sobrevivientes de LLA infantil, la leucoencefalopatía aguda durante el tratamiento del SNC con quimioterapia sola pronosticó riesgos más altos de problemas neuroconductuales a largo plazo (por ejemplo, deficiencias en la organización e iniciación de tareas [componentes del funcionamiento ejecutivo]) y reducción de la integridad de la sustancia blanca en las regiones frontales del encéfalo.[62]
En comparación con la irradiación craneal, la quimioterapia sola dirigida al SNC produce deficiencias neurocognitivas que comprometen los procesos de atención, la velocidad de procesamiento de la información, la memoria, la comprensión verbal, las destrezas visoespaciales, el funcionamiento visomotriz y el funcionamiento ejecutivo; aunque se suele preservar el funcionamiento intelectual general.[56,60,63-65]
Se dispone de pocos estudios longitudinales en los que se evalúen los desenlaces neurocognitivos a largo plazo y que notifiquen datos adecuados sobre el deterioro del CI general después del tratamiento con quimioterapia sola.[63]
Los logros académicos a largo plazo de los sobrevivientes de LLA se ubican en general en el promedio de lectura y ortografía; las deficiencias afectan sobre todo el desempeño en aritmética.[60,66]
Los factores de riesgo de un desenlace neurocognitivo adverso después del tratamiento con quimioterapia sola dirigida al SNC son la edad temprana y el sexo femenino.[65,67]
Se ha observado un estado de limitación cognitiva relacionado con una integridad reducida de las regiones neuroanatómicas esenciales para la formación de recuerdos (por ejemplo, volumen hipocámpico disminuido con aumento de la activación y cortezas parietales más delgadas).[62]
Todavía se investiga de forma activa el efecto a largo plazo de estas anomalías neurocognitivas y de neuroimágenes prevalentes en el estado funcional en adultos en proceso de envejecimiento tratados por una LLA infantil, en particular, aquellos tratados con abordajes contemporáneos de quimioterapia sola.
La investigación en sobrevivientes de LLA infantil que recibieron tratamiento con quimioterapia sola indica que la predisposición genética tal vez module el riesgo de presentar efectos neurocognitivos tardíos. También proporciona información sobre los posibles mecanismos subyacentes a las deficiencias neurocognitivas.[68,69]
Evidencia (funcionamiento neurocognitivo de cohortes grandes de sobrevivientes de cáncer infantil):
Los investigadores del CCSS identificaron asociaciones específicas por sexo entre el tratamiento y las afecciones crónicas con las alteraciones neurocognitivas en 1207 sobrevivientes de LLA (media de edad, 30,6 años) tratados con quimioterapia sola.[7]
Los sobrevivientes de ambos sexos notificaron aumento de la prevalencia de alteración de la eficiencia de tareas (atención y velocidad de procesamiento; oportunidad relativa ajustada [OR], 1,89 para hombres; OR, 1,50 para mujeres) y deterioro de la memoria (OR, 1,89 para hombres; OR, 1,96 para mujeres) en comparación con 2273 hermanos o hermanas del mismo sexo.
Entre los varones sobrevivientes, la alteración en la eficiencia de tareas se asoció con afecciones neurológicas de grado 2 a 4 (OR, 4,33) y afecciones pulmonares (OR, 4,99). La alteración en la memoria se asoció con un aumento de la dosis acumulada de metotrexato intratecal (OR, 1,68) y exposición a dexametasona (OR, 2,44).
En las mujeres sobrevivientes, las afecciones endocrinas de grado 2 a 4 se asociaron con riesgo más alto de alteraciones en la eficiencia de tareas (OR, 2,19) y memoria (OR, 2,26).
En el CCSS se examinaron los problemas cognitivos, de comportamiento y aprendizaje, notificados por los padres de 1560 adolescentes sobrevivientes de LLA infantil que se trataron con quimioterapia sola entre 1970 y 1999.[70]
Los sobrevivientes tratados con irradiación craneal tuvieron una frecuencia mucho más alta de problemas en ansiedad y depresión, desatención e hiperactividad, así como retraimiento social que los pacientes que no recibieron irradiación craneal.
En comparación con los hermanos, los sobrevivientes tratados con quimioterapia sola tuvieron más probabilidad de demostrar comportamiento obstinado (19 de los sobrevivientes vs. 14 % de los hermanos, P = 0,010), desatención e hiperactividad (19 vs. 14 %, P < 0,0001), retraimiento social (18 vs. 12 %, P = 0,002) y presentaron tasas más altas de problemas de aprendizaje (28 vs. 14 %, P < 0,0001).
En modelos multivariantes aplicados a los sobrevivientes, el aumento de la dosis acumulada de metotrexato intravenoso (es decir, >4,3 g/m2) confirió un aumento de riesgo de desatención e hiperactividad (RR, 1,53).
Los sobrevivientes adolescentes con problemas cognitivos o de comportamiento, y aquellos con problemas de aprendizaje tuvieron menos probabilidades de graduarse en la universidad en la juventud que los sobrevivientes adolescentes sin esos problemas.
Los problemas de desatención e hiperactividad se relacionaron con el riesgo más alto de recibir educación especial durante la adolescencia. La participación en educación especial durante la adolescencia no mejoró los logros educativos en la edad adulta.
En el ensayo del SJCRH TOTAL XVI (NCT00549848) se evaluó si una dosis más alta de pegaspargasa y la intensificación temprana de la terapia intratecal triple (TIT; metotrexato, hidrocortisona y citarabina) mejorarían el control sistémico y del SNC en niños con LLA recién diagnosticada. La TIT intensificada se definió como más de 21 dosis acumuladas para pacientes con LLA de riesgo bajo (70 de 194) y más de 27 dosis para aquellos con LLA de riesgo estándar y alto (81 de 206). Las pruebas neurocognitivas se completaron al final del tratamiento.[71]
El grupo general presentó normas por debajo de la edad en las medidas de inteligencia global (P < 0,0001), atención (P = 0,0051), memoria operativa (P = 0,0001), velocidad de procesamiento (P = 0,0002), velocidad motora fina (P = 0,0001) y habilidades matemáticas (P = 0,0087). Las calificaciones del cuidador del funcionamiento del paciente mostraron riesgos elevados de problemas de atención P = 0,0173), función ejecutiva (P = 0,0001) y habilidades de adaptación (P = 0,0001).
En el grupo de riesgo bajo, no hubo diferencias significativas entre los pacientes tratados con ITT intensificada o sin esta.
Los pacientes con LLA de riesgo estándar y alto que recibieron tratamiento con ITT intensificada presentaron una memoria operativa más precaria (P = 0,0328) y velocidad motora fina (P = 0,0403), y índices elevados de falta de atención (P = 0,0189) y disfunción ejecutiva (P = 0,0245). Las mujeres de este grupo de riesgo tratadas con ITT intensificada presentaron puntuaciones de memoria operativa más bajas.
En el ensayo del SJCRH Total XV (NCT00137111), en el que se omitió la irradiación craneal profiláctica, la evaluación cognitiva integral realizada en la semana 120 a 243 pacientes reveló lo siguiente:[72]
Un riesgo más alto de funcionamiento inferior al promedio en la medición de la atención sostenida, pero no en la del funcionamiento intelectual, las habilidades académicas o la memoria.
El riesgo de deficiencias cognitivas se correlacionó con la intensidad del tratamiento, pero no con la edad ni el sexo en el momento del diagnóstico.
El seguimiento prolongado (promedio de 7,7 años desde el diagnóstico) de esta cohorte permitió demostrar que la inteligencia estaba dentro de los límites normales en comparación con las expectativas poblacionales, pero las mediciones del funcionamiento ejecutivo, la velocidad de procesamiento y la memoria estuvieron por debajo de las medias poblacionales.
Las concentraciones sanguíneas más altas de metotrexato se relacionaron con disfunción ejecutiva, corteza cerebral más gruesa y actividad más alta de las regiones frontales del encéfalo en la IRM funcional.
En un estudio prospectivo grande sobre desenlaces neurocognitivos en niños con LLA recién diagnosticada, se asignó al azar a 555 niños a recibir terapia dirigida al SNC según el grupo de riesgo.[73]
El grupo de riesgo bajo se asignó al azar a recibir metotrexato intratecal o dosis altas de metotrexato.
El grupo de riesgo alto se asignó al azar a recibir dosis altas de metotrexato o 24 Gy de radioterapia craneal.
Se observó una reducción significativa en las puntuaciones de CI (4–7 puntos) en todos los grupos de pacientes cuando se compararon con los controles, con independencia del tratamiento administrado al SNC.
Fue más probable que los niños menores de 5 años en el momento del diagnóstico tuvieran CI inferiores a 80 al cabo de 3 años del tratamiento que los niños mayores de 5 años en el momento del diagnóstico, con independencia del tratamiento asignado, lo cual indica que los niños más pequeños son más vulnerables a los efectos tóxicos neurológicos del tratamiento.
Se han observado deficiencias cognitivas persistentes y deterioro intelectual progresivo en cohortes de adultos tratados por LLA en la niñez que se relacionan con una reducción de los logros educativos y desempleo.[5,55,59] Los resultados de un estudio de más de 500 adultos sobrevivientes de LLA infantil (promedio de 26 años después del diagnóstico) mostraron lo siguiente:[55]
Los sobrevivientes exhibieron mayores tasas de deterioro en todos los dominios neurocognitivos (oscilaron entre un 28,6 % y un 58,9 % en cada dominio).
La tasa de deterioro grave aumentó en función de la dosis de radiación craneal, pero fue común entre los sobrevivientes tratados con dosis más bajas de radiación craneal y quimioterapia sola.
El deterioro de las habilidades del funcionamiento ejecutivo aumentó con el tiempo a partir del diagnóstico según la dosis de radiación craneal; el deterioro intelectual, académico y de la memoria aumentó de forma progresiva a medida que bajaba la edad en el momento de administración de la radiación craneal y según la dosis; y el deterioro neurocognitivo se relacionó con desenlaces funcionales en la edad adulta, incluso menos probabilidades de obtener un título universitario y un empleo a tiempo completo.
Leucemia linfoblástica aguda y terapia con corticoesteroides
El tipo de corticoesteroide que se usa para el tratamiento sistémico de la LLA quizás afecte el funcionamiento cognitivo.
En un estudio que incluyó pruebas neurocognitivas a largo plazo (media de seguimiento de 9,8 años) realizadas a 92 niños con antecedentes de LLA de riesgo estándar que recibieron dexametasona o prednisona durante el tratamiento, no se observaron diferencias significativas en las puntuaciones medias del rendimiento neurocognitivo y académico.[74]
En contraste, en un estudio de 567 adultos sobrevivientes de leucemia infantil (media de edad, 33 años; media de tiempo desde el diagnóstico, 26 años), se notificaron los siguientes resultados:[55]
La exposición a la dexametasona se asoció con un aumento del riesgo de alteración en la memoria (RR, 2,12; intervalo de confianza [IC] 95 %, 1,11–4,03) y el funcionamiento ejecutivo (RR, 2,42; IC 95 %, 1,20–4,91), de manera independiente de la exposición al metotrexato.
La hidrocortisona intratecal también aumentó el riesgo de problemas de atención (RR, 1,24; IC 95 %, 1,05–1,46).
Otros tipos de cáncer
Se notificaron anomalías neurocognitivas en otros grupos de sobrevivientes de cáncer. En un estudio de adultos sobrevivientes de cánceres infantiles fuera del SNC (entre ellos, LLA, n = 5937) se notificaron los siguientes resultados:[57]
Del 13 % al 21 % de los sobrevivientes notificaron alteraciones en la eficiencia de tareas, organización, memoria o regulación emocional. Esta tasa de deterioro fue casi un 50 % más alta que la notificada en el grupo de comparación de hermanos.
Los factores relacionados con el deterioro fueron un diagnóstico anterior a los 6 años de edad, sexo femenino, radioterapia craneal y deterioro de la audición.
Datos recientes indican que las afecciones crónicas que se inician durante la edad adulta tal vez contribuyan a los deterioros cognitivos en los sobrevivientes a largo plazo de cánceres fuera del SNC.
En un análisis del estudio de cohorte SJLIFE se evaluó si los niños que presentaron lesiones en el SNC tenían un riesgo más alto de deterioro neurocognitivo relacionado con afecciones crónicas de inicio tardío subsiguientes. Un grupo de 2859 sobrevivientes de 18 años o más completaron un conjunto de análisis neurocognitivo y un examen clínico al cabo de, por lo menos, 10 años del diagnóstico. De estos pacientes, 1598 había recibido tratamiento dirigido al SNC, como radiación craneal, metotrexato intratecal o neurocirugía.[75]
Los sobrevivientes que recibieron tratamiento en el SNC tuvieron un desempeño más deficiente en todas las pruebas neurocognitivas en comparación con los sobrevivientes que no recibieron tratamiento dirigido al SNC, y fue más probable que presentaran deterioro neurocognitivo general (46,9 vs. 35,3 %).
Se observó una relación entre la dosis y la respuesta y la puntuación de gravedad o entre la carga de afecciones crónicas y el deterioro neurocognitivo solo entre los sobrevivientes que recibieron tratamiento dirigido al SNC (OR alto, 2,24; IC 95 %, 1,42–3,53; OR muy alto, 4,07; IC 95 %, 2,30–7,17).
Se estableció una relación entre las afecciones cardiovasculares y pulmonares y los desenlaces cognitivos (velocidad de procesamiento, funcionamiento ejecutivo y deterioro de la memoria) solo en los sobrevivientes con afecciones neurológicas que habían recibido tratamiento dirigido al SNC.
Se notificaron anomalías neurocognitivas como resultado de los siguientes cánceres:
Leucemia mieloide aguda (LMA). Investigadores del CCSS compararon los desenlaces neurocognitivos y psicosociales de 482 sobrevivientes (mediana de edad, 30 años) con los de 3190 hermanos del grupo de control (mediana de edad, 32 años).[76]
En comparación con los hermanos, los sobrevivientes de LMA fueron más propensos a notificar deterioro de la calidad de vida emocional general (CRI, 2,19; IC 95 %, 1,51–3,18), neurocognitiva (CRI, 2,03; IC 95 %, 1,47–2,79) y física (CRI, 2,71; IC 95 %, 1,61–4,56).
Los sobrevivientes también tenían un aumento del riesgo de deficiencias psicosociales, incluso un menor nivel de educación (CRI, 1,15; IC 95 %, 1,03–1,30), desempleo (CRI, 1,41; IC 95 %, 1,16–1,71) e ingresos más bajos (CRI, 1,39; IC 95 %, 1,17–1,65).
Los desenlaces no fueron estadísticamente diferentes entre los sobrevivientes que recibieron un trasplante y los sobrevivientes que solo recibieron quimioterapia intensiva.
Osteosarcoma. En un estudio, en el que se evaluó el funcionamiento neurocognitivo de 80 sobrevivientes a largo plazo de osteosarcoma (media de tiempo desde el diagnóstico, 24,7 años), los sobrevivientes presentaron puntuaciones medias más bajas en las habilidades de lectura, atención, memoria y velocidad de procesamiento que los controles comunitarios.[77]
La presencia de afecciones cardíacas, pulmonares y endocrinas exhibió una relación significativa con un desempeño más deficiente en las mediciones de memoria y velocidad de procesamiento.
Sarcoma de tejido blando. En un informe del CCSS se caracterizó la prevalencia y los factores de riesgo de deterioro neurocognitivo autonotificado en sobrevivientes de rabdomiosarcoma (n = 713) y en sus hermanos (n = 706). También se identificaron los factores de riesgo relacionados con estos desenlaces en los sobrevivientes.[78]
Más sobrevivientes notificaron deficiencias neurocognitivas, en comparación con sus hermanos (eficiencia de tareas: 21,1 vs. 13,7 %; regulación emocional: 16,7 vs. 11,0 %; memoria: 19,3 vs. 15,1 %). La exposición a la radiación craneal aumentó el riesgo de deterioro de la eficiencia de tareas (OR, 2,30).
El consumo de tabaco se relacionó con un deterioro de la eficiencia de tareas (OR, 2,06) y de la memoria (OR, 2,23).
Las afecciones neurológicas y auditivas se asociaron con alteraciones de la memoria (OR, 2,44 y OR, 1,87, respectivamente).
Los investigadores del estudio SJLIFE evaluaron el funcionamiento neurocognitivo y el estado de salud de 150 sobrevivientes de sarcoma de tejido blando infantil (mediana de edad, 33 años; mediana de tiempo desde el diagnóstico, 24 años) mediante evaluaciones clínicas objetivas.[79]
En comparación con los controles comunitarios y la población, los sobrevivientes presentaron parámetros más bajos de razonamiento verbal, matemáticas y memoria a largo plazo.
Se observó que la exposición acumulada a las antraciclinas (cada 100 mg/m2) se relacionaba con valores de razonamiento verbal, lectura y vitalidad notificada por el paciente más precarios.
Las afecciones neurológicas y neurosensoriales crónicas se relacionaron con puntuaciones más precarias en el desempeño matemático e hipoacusia.
Un mejor rendimiento cognitivo se relacionó con un mayor logro social.
Tumor de Wilms. Los investigadores del estudio SJLIFE examinaron la prevalencia y los predictores de desenlaces neurocognitivos, logro social, sufrimiento emocional y calidad de vida relacionada con la salud en los sobrevivientes a largo plazo de tumor de Wilms (n = 158; mediana de tiempo desde el diagnóstico, 29 años; mediana de edad, 33 años) y compararon los resultados con controles comunitarios.[80]
Los sobrevivientes a largo plazo de tumor de Wilms presentaron un funcionamiento neurocognitivo más bajo, que abarcó el razonamiento verbal, el desempeño académico, la atención, la memoria y el funcionamiento ejecutivo, en comparación con los controles.
Fue menos probable que los sobrevivientes de tumor de Wilms se graduaran de la universidad (OR, 2,23) y presentaran más afecciones neurológicas de moderadas a graves en comparación con los controles (18,4 vs. 8,2 %; P < 0,001).
Retinoblastoma. La evaluación en serie del funcionamiento cognitivo y adaptativo en un grupo de sobrevivientes menores de 6 años reveló deterioro del desarrollo funcional a lo largo del tiempo; el deterioro más pronunciado se observó en pacientes con deleciones 13q.[81,81]
En un seguimiento longitudinal posterior de esta cohorte se identificaron mejoras del funcionamiento adaptativo en todos los grupos de tratamiento y en el funcionamiento cognitivo de los sobrevivientes tratados con enucleación sola entre los 5 y 10 años de edad.[82]
A los 10 años de edad, el funcionamiento general se mantuvo en el intervalo promedio, si bien el CI calculado fue significativamente inferior a la media normativa para los niños tratados con enucleación sola.[82]
En un estudio de adultos sobrevivientes a muy largo plazo, en promedio 33 años después del diagnóstico, se demostró, en general, un funcionamiento cognitivo dentro del promedio para los dominios de inteligencia, memoria, atención y funcionamiento ejecutivo.[83]
Linfoma. En el pasado, los sobrevivientes de linfoma no se consideraban en riesgo de presentar efectos tardíos neurocognitivos.
Sin embargo, en un informe se observó que más de dos tercios de los sobrevivientes de linfoma no Hodgkin infantil experimentaron por lo menos un leve deterioro neurocognitivo, incluso alteraciones graves en el funcionamiento ejecutivo (13 %), la atención (9 %) y la memoria (4 %).[84]
De manera similar, en un estudio de 62 adultos sobrevivientes de linfoma de Hodgkin infantil, los sobrevivientes tuvieron un peor desempeño en las mediciones de atención sostenida, memoria a corto y largo plazo, y fluidez cognitiva en comparación con datos normativos nacionales.[85] Cabe destacar que las mediciones del funcionamiento cardíaco y pulmonar también se vincularon con deterioro neurocognitivo en este grupo de sobrevivientes.
Neuroblastoma. Los investigadores del CCSS compararon los desenlaces cognitivos entre 837 sobrevivientes de neuroblastoma (mediana de edad, 1 año en el momento del diagnóstico; mediana de edad alcanzada, 25 años) y una cohorte de hermanos (mediana de edad alcanzada, 23 años) mediante el CCSS NeuroCognitive Questionnaire (NCQ).[86]
Los sobrevivientes tuvieron un riesgo un 50 % mayor de deterioro en la atención y la velocidad de procesamiento, así como en la regulación emocional.
El riesgo de deterioro neurocognitivo fue más alto en los sobrevivientes con afecciones crónicas de salud.
También fue menos probable que los sobrevivientes alcanzaran hitos de la vida adulta, como vivir de manera independiente.
Trasplante de células madre hematopoyéticas
También se evaluaron las consecuencias cognitivas y académicas de un trasplante de células madre hematopoyéticas (TCMH) en niños, y estas incluyen, entre otras, las siguientes:
En un estudio de encuesta se evaluaron los resultados neurocognitivos tardíos entre 199 sobrevivientes de TCMH (TCMH a la edad de <21 años; mediana de seguimiento desde el TCMH, 27,6 años).[87]
Según la evaluación del CCSS NCQ, entre el 18,9 % y el 32,5 % de los sobrevivientes notificaron alteraciones en la eficiencia de las tareas, la memoria, la regulación emocional o la organización, en comparación con la tasa esperada del 10 % (población general).
La calidad de vida, evaluada mediante el Neuro-Quality of Life Cognitive Function Short Form (Neuro-QoL), entre los sobrevivientes (puntuación T media, 49,6) fue comparable a la de la población normativa (50).
Los factores de riesgo independientes de deterioro de la Neuro-QoL (puntuación T <40) incluían problemas auditivos (OR, 4,7; IC 95 %, 1,96–12,6), antecedentes de accidente cerebrovascular o convulsiones (OR, 4,46; IC 95 %, 1,44–13,8) y trastornos del sueño (OR, 6,96; IC 95 %, 2,53–19,1).
Los investigadores del SJCRH examinaron la influencia de la edad y el acondicionamiento con irradiación corporal total (ITC) en la trayectoria de la función cognitiva tras el TCMH. De los 315 pacientes que completaron una evaluación inicial, 183 estaban vivos 1 año después del TCMH y completaron evaluaciones adicionales 1, 3 y 5 años después.[88]
A los 5 años del TCMH, los pacientes más jóvenes (de <3 años de edad al inicio del estudio) que recibieron ICT mostraron un CI significativamente menor que los que no recibieron ICT.
Los análisis longitudinales demostraron un efecto significativo de la edad y la ICT a lo largo del tiempo, con descensos en el funcionamiento cognitivo durante el primer año entre los pacientes más jóvenes y recuperación del funcionamiento en los años posteriores entre los pacientes que no recibieron ICT.
Los pacientes jóvenes que recibieron ICT no recuperaron las pérdidas experimentadas durante el primer año tras el TCMH, demostrando estabilidad en su funcionamiento, pero a un nivel inferior.
Secuelas neurológicas
El riesgo de complicaciones neurológicas quizás esté determinado por los siguientes aspectos:
En los niños con tumores en el SNC, es posible que el efecto de la masa, la infiltración tumoral y el aumento de la presión intracraneal produzcan alteraciones motrices o sensoriales, disfunción cerebelar y efectos secundarios, como convulsiones y complicaciones cerebrovasculares.[89]
En numerosos informes se describen anomalías en la integridad y el funcionamiento del SNC, pero tales estudios suelen tener limitaciones por el tamaño pequeño de la muestra, el sesgo de selección de la cohorte y el sesgo de participación, la determinación transversal de los resultados y el período variable de tiempo de evaluación desde las exposiciones del tratamiento. Por el contrario, en relativamente pocos estudios se determinan de modo integral o sistemático los desenlaces relacionados con el funcionamiento del sistema nervioso periférico.
Los sobrevivientes de tumores del SNC siguen teniendo un riesgo más alto de nuevas complicaciones neurológicas adversas a lo largo de la vida en comparación con sus hermanos. No se ha alcanzado ninguna meseta para las nuevas secuelas adversas, incluso 30 años después del diagnóstico, según un estudio longitudinal de 1876 sobrevivientes de tumores del SNC a 5 años del CCSS. La mediana de tiempo desde el diagnóstico fue de 23 años y la mediana de edad de los pacientes estudiados fue de 30,3 años.[90]
La radiación craneal, el accidente cerebrovascular, la recidiva del tumor y la aparición de un meningioma se relacionaron de modo independiente con un inicio tardío de secuelas neurológicas (convulsiones, disfunción neurológica focal y anomalías neurosensoriales).
Este hallazgo apoya la necesidad de controlar en forma minuciosa a estos pacientes con seguimiento neurológico continuo en el marco o en estrecha asociación con un centro de atención multidisciplinaria para sobrevivientes de cáncer.
Las complicaciones neurológicas que a veces se presentan en los adultos sobrevivientes de cáncer infantil son las siguientes:
Convulsiones
Las convulsiones pueden ser secundarias al efecto de la masa tumoral dentro del SNC o por terapias neurotóxicas dirigidas al SNC.
En 1876 sobrevivientes a 5 años de tumores del SNC del estudio CCSS, la incidencia de convulsiones aumentó del 27 % en sobrevivientes a 5 años desde el diagnóstico hasta el 41 % en sobrevivientes a 30 años desde el diagnóstico.[90]
Las convulsiones de inicio tardío se relacionaron con radiación de 50 Gy dirigida al lóbulo frontal (cociente de riesgos instantáneos [CRI], 1,8) y radiación dirigida al lóbulo temporal de manera dependiente de la dosis (CRI, 1,9 para 1–49 Gy; CRI, 2,2 para >50 Gy).
Otros factores de riesgo relacionados con convulsiones de inicio tardío incluyeron recidiva (CRI, 2,3), presentación de un meningioma (CRI, 2,6) y antecedentes de accidente cerebrovascular (CRI, 2,0).
El riesgo de convulsiones fue más elevado en los sobrevivientes que en los hermanos (CRI, 12,7).
Entre los sobrevivientes de leucemia infantil en el CCSS (n = 4151; 64,5 % tratados con irradiación craneal), el 6,1 % notificó un trastorno convulsivo, y las convulsiones ocurrieron más de 5 años después del diagnóstico en el 51 % de estos pacientes.[91]
En un análisis del estudio SJLIFE se evaluó el efecto de los factores relacionados con las convulsiones sobre los desenlaces neurocognitivos, la calidad de vida relacionada con la salud (CVRS) y sociales en 2022 sobrevivientes de cáncer infantil (mediana de edad, 31,5 años; mediana de tiempo desde el diagnóstico, 23,6 años).[92]
Se identificaron convulsiones en 232 sobrevivientes (el11,5 %; el 29,9 % de los sobrevivientes con tumores del SNC y el 9,0 % de los sobrevivientes sin tumores del SNC).
Entre los sobrevivientes de tumores del SNC, las convulsiones se relacionaron con una función ejecutiva y una velocidad de procesamiento más precarios (según las puntuaciones z estándar ajustadas por edad). La resolución de las convulsiones se relacionó con una mejora de la atención y la memoria.
Entre los sobrevivientes de tumores que no afectan el SNC, las convulsiones se relacionaron con una peor función en todos los dominios cognitivos, en comparación con los sobrevivientes sin convulsiones. La gravedad de las convulsiones se relacionó con una peor velocidad de procesamiento, y la resolución de las convulsiones se relacionó con una mejor función ejecutiva y atención.
Los sobrevivientes de tumores fuera del SNC con convulsiones tuvieron desenlaces más precarios de CVRS (funcionamiento físico, limitaciones de tareas físicas y salud general). Las convulsiones, tanto en los sobrevivientes de tumores del SNC como en los sobrevivientes de tumores fuera del SNC, aumentaron el riesgo de desempleo y de empleo a tiempo parcial.
Neuropatía periférica
Los alcaloides de vinca (vincristina y vinblastina) y los metales pesados (cisplatino y carboplatino) pueden causar neuropatía periférica.[93-95]
La neuropatía periférica se presenta durante el tratamiento y suele mejorar o resolverse, desde el punto de vista clínico, después del tratamiento.[93] Sin embargo, en estudios recientes de sobrevivientes a largo plazo se indica que es probable que no se haya evaluado lo suficiente la neuropatía periférica inducida por quimioterapia.[96]
Dosis más altas acumuladas de vincristina o metotrexato intratecal se relacionaron con deficiencias neuromusculares en los sobrevivientes a largo plazo de LLA infantil, lo cual indica que los efectos persistentes de estos fármacos afectan el estado funcional de los sobrevivientes que envejecen.[93]
Los investigadores del estudio SJLIFE encontraron vínculos entre la neuropatía periférica y el deterioro en las mediciones de desempeño del movimiento (movilidad y resistencia al caminar) y la calidad de vida (funcionamiento físico, desempeño físico y salud general) entre los sobrevivientes de LLA infantil.[97]
La alteración del equilibrio estático de pie (factor de predicción de caída en los siguientes 90 días) fue más frecuente en los sobrevivientes en comparación con los controles, pero no se relacionó con la neuropatía periférica.[97]
En los adultos sobrevivientes de tumores extracraneales sólidos en la niñez (mediana de tiempo desde el diagnóstico de 25 años), la evaluación estandarizada del funcionamiento neuromuscular reveló insuficiencias motoras relacionadas con la exposición a la vincristina y deterioro sensorial en relación con la exposición al cisplatino.[94]
En los sobrevivientes con discapacidades sensoriales, se observó una prevalencia más alta de limitaciones en el desempeño funcional, relacionadas con restricciones en la capacidad de resistencia y movilidad.
Estos estudios ponen de relieve la importancia de la evaluación y la derivación a servicios de rehabilitación para optimizar los desenlaces funcionales en los sobrevivientes a largo plazo.
Accidente cerebrovascular u otros efectos cerebrovasculares
Los sobrevivientes de tumores del SNC en la niñez tienen un riesgo 43 veces más elevado de sufrir accidentes cerebrovasculares en comparación con los hermanos.[43,98]
La radioterapia craneal (dependiente de la dosis), la arteroesclerosis inicial, la hipertensión y la etnia afroamericana son factores de riesgo identificados.[99-101] Para obtener información sobre el accidente cerebrovascular, consultar la sección Enfermedad cerebrovascular.
En el CSS británico (n = 13 457 sobrevivientes), en el que participaron 2885 sobrevivientes de tumores del SNC infantil, se notificaron los siguientes resultados:[102]
Los sobrevivientes de tumores del SNC que recibieron irradiación craneal presentaron aumento del riesgo de enfermedad cardiovascular entre los 50 y 65 años.
La incidencia acumulada de enfermedad cerebrovascular en sobrevivientes fue del 11,6 % a la edad de 40 años, del 16 % a los 50 años y del 26 % a los 65 años, en comparación con la incidencia esperada del 4,2 %.
Entre los 271 participantes del CCSS con antecedentes de accidente cerebrovascular a una edad mediana de 19 años, 70 notificaron un segundo accidente cerebrovascular a una edad mediana de 32 años.[103]
La incidencia acumulada a 10 años de accidente cerebrovascular recidivante tardío fue del 21 % en total, y del 33 % para los participantes que recibieron tratamiento con radioterapia craneal de 50 Gy o superior.
Los factores de riesgo de accidente cerebrovascular recidivante fueron los siguientes:
Radioterapia craneal de 50 Gy o superior (CRI, 4,4; IC 95 %, 1,4–13,7).
Hipertensión (CRI, 1.9; IC 95 %, 1,0–3.5).
Edad avanzada en el momento del primer accidente cerebrovascular (CRI, 6.4; IC 95 %, 1,8–23).
En una revisión del PENTEC se identificaron 101 de 3989 pacientes pediátricos que experimentaron al menos un efecto tóxico cerebrovascular (accidente isquémico transitorio, accidente cerebrovascular, moyamoya o arteriopatía).[104]
Las incidencias previstas se basaron en la dosis de radiación dirigida al polígono de Willis: 0,2 % a 30 Gy, 1,3 % a 45 Gy y 4,4 % a 54 Gy.
A una edad alcanzada de 35 años, la incidencia de accidente cerebrovascular prevista fue del 0,9 % al 1,3 % para 30 Gy, del 1,8 % al 2,7 % para 45 Gy y del 2,8 % al 4,1 % para 54 Gy (en comparación con un riesgo basal de 0,2–1,0 %).
A la edad de 45 años, la incidencia prevista de accidente cerebrovascular fue del 1,2 % al 4,2 % para 30 Gy, del 4,5 % al 8,6 % para 45 Gy y del 6,7 % al 13 % para 54 Gy (en comparación con un riesgo basal del 0,5–1,0 %).
Hipersomnia (somnolencia diurna) o narcolepsia
En una revisión retrospectiva de pacientes con tumor de encéfalo tratados en el SJCRH, los investigadores identificaron a 39 de 2336 pacientes con diagnóstico de hipersomnia o narcolepsia, para una tasa de prevalencia de 1670 casos por 100 000, muy por encima de una tasa de prevalencia de 20 a 50 casos por 100 000 notificados en la población general.[105]
Esto podría ser una subestimación en los sobrevivientes de tumores de encéfalo infantiles porque en muchos pacientes con síntomas leves a moderados, como fatiga y trastornos del sueño, a veces no se reconocen estos trastornos ni se derivan a un especialista del sueño.
La hipersomnia o narcolepsia se diagnosticó a una mediana de 6 años (intervalo, 0,4–13,2 años) desde el diagnóstico del tumor y 4,7 años (intervalo, 1,5–10,4 años) desde la radiación craneal.
La ubicación del tumor en la línea media y el uso de medicamentos antiepilépticos se correlacionaron con hipersomnia o narcolepsia, mientras que una dosis de radiación superior a 30 Gy tendió a ser significativa.
La ubicación del tumor en la fosa posterior se relacionó con un riesgo reducido de hipersomnia.
El tratamiento de la hipersomnia o narcolepsia se debe individualizar y la intervención farmacológica con estimulantes a veces resulta beneficiosa.
En una evaluación inicial de 82 sobrevivientes de tumores del SNC infantil (mediana de edad, 13,8 años) que participaban en un ensayo controlado aleatorizado sobre neurorretroalimentación, el 48 % de los sobrevivientes refirieron problemas para dormir y obtuvieron puntuaciones significativamente peores que la norma en la Sleep Disturbance Scale for Children en las subescalas de inicio y mantenimiento del sueño, de somnolencia excesiva y en la escala total.[106]
Los problemas emocionales y la hiperactividad o desatención fueron posibles factores de riesgo independientes para los problemas para dormir. Los problemas para dormir se relacionaron también con un peor funcionamiento ejecutivo notificado por los progenitores.
Otras secuelas neurológicas
En un informe del CCSS en el que se compararon los efectos tardíos neurológicos autonotificados de 4151 adultos sobrevivientes de LLA infantil con sus hermanos, los sobrevivientes tuvieron un riesgo elevado de inicio tardío de problemas de coordinación, problemas motrices, convulsiones y cefaleas.[91]
La incidencia general acumulada fue del 44 % a los 20 años. Las cefaleas intensas fueron las más frecuentes, con una incidencia acumulada del 25,8 % a los 20 años, seguida de disfunción neurológica focal (21,2 %) y convulsiones (7 %).
Los niños tratados con regímenes que incluían irradiación craneal para la LLA y los que presentaron una recaída tuvieron un aumento del riesgo de secuelas neurológicas de inicio tardío.
En un estudio transversal se evaluó la morbilidad neurológica y la calidad de vida de 162 sobrevivientes de LLA infantil (mediana de edad en el momento de la evaluación, 15,7 años; mediana de edad desde el momento de finalización del tratamiento, 7,4 años) y además se sometieron a un examen neurológico.[107]
Se presentaron síntomas neurológicos en el 83 % de los sobrevivientes, pero la morbilidad relacionada con los síntomas fue baja y la calidad de vida fue alta en la mayoría de los sobrevivientes.
Los síntomas notificados con más frecuencia fueron neuropatía (63 %), cefalea (46,9 %), mareos (33,3 %) y dorsalgia (22,8 %).
El sexo femenino, haber recibido 10 dosis o más de quimioterapia intratecal, la irradiación craneal, la leucemia en el SNC en el momento del diagnóstico y los antecedentes de recaída de la LLA se relacionaron con morbilidad neurológica.
Los estudios de neuroimagen en sobrevivientes de LLA que recibieron irradiación y aquellos que no la recibieron exhiben una variedad de anomalías del SNC, como leucoencefalopatía, lagunas cerebrales, atrofia cerebral y calcificaciones distróficas (microangiopatía mineralizante).[53,59,108,109]
Entre estas, las anomalías en la integridad y el volumen de la sustancia blanca cerebral se correlacionaron con los desenlaces neurocognitivos.
También se observaron cavernomas en sobrevivientes de LLA infantil tratados con irradiación craneal. Se ha especulado que son el resultado de procesos angiogénicos en oposición a oncogénicos.[110]
En un estudio transversal retrospectivo, 101 sobrevivientes de LLA infantil (media de tiempo desde el diagnóstico, 27,6 años) tratados con irradiación craneal (63,4 % recibieron ≤18 Gy) se sometieron a pruebas neurocognitivas e IMR tridimensional del encéfalo.[111]
Mediante el uso de imágenes ponderadas de susceptibilidad se identificaron hemorragias intracerebrales focales pequeñas que solo son visibles con secuencias de IRM muy sensibles.
Se encontró por lo menos 1 microhemorragia en el 85 % de los sobrevivientes, y el sitio de presentación más frecuente fueron los lóbulos frontales.
Una dosis de radiación de 24 Gy produjo un riesgo 5 veces más alto de presentar múltiples microhemorragias, en comparación con una dosis de 18 Gy.
No se encontraron diferencias significativas en las puntuaciones neurocognitivas con la presencia o ausencia de microhemorragias ni su ubicación.
Los investigadores del CCSS evaluaron las secuelas neurológicas relacionadas con el tratamiento en sobrevivientes de tumores del SNC infantil.[90]
En 1876 sobrevivientes a 5 años de tumores del SNC del estudio CCSS, la incidencia acumulada de cefaleas aumentó desde el 38 % a los 5 años del diagnóstico hasta el 53 % a 30 años del diagnóstico.
Los problemas de coordinación aumentaron del 21 % 5 años después del diagnóstico, hasta el 53 % 30 años después del diagnóstico; las alteraciones motoras aumentaron del 21 % hasta el 35 % durante este período.
El aumento de riesgo de alteraciones motoras se relacionó con la recidiva tumoral (CRI, 2,6), presentación de un meningioma (CRI, 2,3) y accidente cerebrovascular (CRI, 14,9).
La incidencia acumulada de hipoacusia aumentó del 9 % 5 años después del diagnóstico, al 23 % 30 años después del diagnóstico; la incidencia acumulada de acúfenos aumentó del 8 % 5 años después del diagnóstico, al 21 % 30 años después del diagnóstico; y la incidencia acumulada de vértigo aumentó del 9 % 5 años después del diagnóstico, al 17 % 30 años después del diagnóstico.
Los riesgos de deterioro motor (CRI, 7,6) e hipoacusia (CRI, 18,4) fueron elevados, en comparación con los riesgos de los hermanos.
Los investigadores del CCSS calcularon la prevalencia y la incidencia acumulada de la disfunción neuromuscular (disfunción motora o sensorial) entre 25 583 sobrevivientes de cáncer infantil y 5044 hermanos.[112]
La disfunción neuromuscular fue prevalente en el 14,7 % de los sobrevivientes 5 años después del diagnóstico, en comparación con el 1,5 % en los hermanos (razón de prevalencia [RP], 9,9 %; IC 95 %, 7,9–12,4).
La prevalencia de la disfunción neuromuscular fue mayor en los sobrevivientes de tumores del SNC (RP, 27,6; IC 95 %, 22,1–34,6) y de sarcomas (RP, 11,5; IC 95 %, 9,1–14,5).
La incidencia acumulada a 20 años aumentó hasta el 24,3 % en los sobrevivientes 20 años después del diagnóstico.
Los tratamientos contra el cáncer relacionados con el aumento de la prevalencia de la disfunción neuromuscular incluyeron la radioterapia vertebral, el aumento de las dosis de radioterapia craneal y los derivados del platino.
La disfunción neuromuscular se relacionó con resultados adversos posteriores, como obesidad concurrente o posterior (RP, 1,1; IC 95 %, 1,1-1,2), ansiedad (RP, 2,5; IC 95 %, 2,2–2,9), depresión (RP, 2,1; IC 95 %, 1,9–2,3) y menor probabilidad de graduarse en la universidad (RP, 0,92; IC 95 %, 0,90–0,94) y de encontrar empleo (RP, 0,8; IC 95 %, 0,8–0,9).
Los investigadores de SJLIFE evaluaron el deterioro motor y sensorial en 378 sobrevivientes de tumores infantiles del SNC (5 años o más desde el diagnóstico) y 445 controles emparejados por edad, sexo y raza, mediante la Total Neuropathy Score.[113]
Entre los sobrevivientes, 91 (24,1 %; IC 95 %, 19,8–29,4 %) tenían alguna discapacidad motora o sensorial de grado 2 o superior (solo motora, 27 [7,1 %]; solo sensorial, 40 [10,6 %]; ambas, 24 [6,4 %]).
Los sobrevivientes tenían mayor propensión que los controles a presentar un deterioro de grado 2 o superior (13,5 % para los sobrevivientes [7,7 % para el grado 2 y 5,8 % para el grado 3] vs. 0,9 % para los controles; P < 0,001).
Los sobrevivientes de ependimoma presentaban la mayor prevalencia de discapacidad motora (22 %) y los sobrevivientes de meduloblastoma tenían la mayor prevalencia de discapacidad sensorial (33,3 %).
Los factores de tratamiento relacionados con el deterioro motor de grado 2 o superior en los modelos multivariables incluyeron la exposición a alcaloides de vinca de 15 mg/m2 o menos, en comparación con ninguna exposición (OR, 4,38; IC 95 %, 1,06–18,08) y la exposición a etopósido de más de 2036 mg/m2, en comparación con ninguna exposición (OR, 12,61; IC 95 %, 2,19–72,72).
La mielopatía por radiación es una complicación poco frecuente, aunque grave, de la exposición a la radioterapia. El grupo de trabajo sobre médula espinal del PENTEC examinó los factores que contribuyen potencialmente a la mielitis por radiación en la población pediátrica.[114]
Se identificaron 32 pacientes (tratados entre 1962 y 2002) con mielopatía por radiación, con una incidencia bruta del 0,5 %. La mediana de edad fue de 13 años y el 52 % de los pacientes eran hombres. No se identificó ninguna relación aparente con la edad en el momento de la radioterapia. Los diagnósticos de cáncer más frecuentes fueron rabdomiosarcoma, meduloblastoma y linfoma de Hodgkin.
La mediana de la dosis de radiación fue de 40 Gy y el tamaño de la fracción fue de 1,8 Gy. La mediana del período de latencia desde la finalización de la radioterapia hasta la presentación de la mielopatía por radiación fue de 7 meses (intervalo, 1–29 meses) y las dosis de radioterapia más altas se correlacionaron con períodos de latencia más prolongados (P = 0,03).
Las quimioterapias más comunes administradas a pacientes con mielopatía por radiación fueron vincristina, metotrexato intratecal (IT) y citarabina IT. De los pacientes, 10 recibieron vincristina, metotrexato IT y citarabina IT.
La quimioterapia parece reducir la tolerancia de la médula espinal a la radiación. La dosis de radioterapia que produjo mielopatía por radiación fue menor en los pacientes que recibieron quimioterapia que en los que no la recibieron (39,6 ± 8,7 Gy vs. 49,7 ± 8,2 Gy; P = 0,04).
De los 32 casos de mielopatía por radiación, 8 se produjeron en pacientes que se trataron en un único Intergroup Rhabdomyosarcoma Study Group (tratados con quimioterapia intensiva con metotrexato IT y citarabina).
Muchos de los informes databan de una época anterior a la planificación del tratamiento mediante TC. Además, los investigadores no encontraron ningún indicio de que la médula espinal en desarrollo fuera más sensible que la médula espinal adulta.
En el Cuadro 4 se resumen los efectos tardíos en el SNC y los exámenes médicos de detección correspondientes.
Cuadro 4. Efectos tardíos en el sistema nervioso centrala
Tratamiento predisponente
Efectos neurológicos
Exámenes médicos de detección
CI = coeficiente intelectual; IT = intratecal; IV = intravenoso.
Antecedentes: trastornos neurológicos transitorios o permanentes
Evaluación de la presión arterial
Examen neurológico
Neurocirugía encefálica
Alteraciones motoras o sensoriales (parálisis, trastornos del movimiento, ataxia, problemas oculares [parálisis de los nervios oculares, paresia de la mirada, nistagmo, papiledema, atrofia óptica]); convulsiones
Examen neurológico
Evaluación neurológica
Neurocirugía encefálica
Hidrocefalia, disfunción de la derivación
Radiografía del abdomen
Evaluación neuroquirúrgica
Neurocirugía en la columna vertebral
Vejiga neurogénica, incontinencia urinaria
Antecedentes: hematuria, urgencia o frecuencia urinarias, incontinencia o retención urinaria, disuria, nicturia, flujo urinario anormal
Neurocirugía en la columna vertebral
Intestino neurogénico, incontinencia fecal
Antecedentes: estreñimiento crónico e incontinencia fecal
Examen del recto
Tratamiento predisponente
Efectos neurológicos
Exámenes médicos de detección
Metotrexato (dosis alta, IV o IT), citarabina (dosis alta, IV o IT), radioterapia con exposición del encéfalo, neurocirugía encefálica
Deficiencias neurocognitivas (funcionamiento ejecutivo, memoria, atención, velocidad de procesamiento, etc.), deficiencias de aprendizaje, disminución del CI, cambios de comportamiento
Evaluación del progreso educativo y vocacional
Evaluación neuropsicológica formal
Psicosociales
Muchos sobrevivientes de cáncer infantil notifican una reducción de la calidad de vida, deterioro del estado de salud u otros desenlaces psicosociales adversos, en comparación con los grupos de hermanos o de población sin cáncer.[115,116] Se han identificado grupos vulnerables relacionados con factores sociodemográficos (p. ej., sexo femenino), diagnósticos específicos de cáncer (p. ej., tumor del SNC), tratamientos contra el cáncer (p. ej., radioterapia craneal), conductas saludables (p. ej., consumo de tabaco) y tipo o carga acumulada de enfermedades crónicas. El diagnóstico de cáncer infantil a veces afecta los desenlaces psicológicos y el logro de la independencia funcional y social en la vida adulta. En varias investigaciones se demostró que los sobrevivientes de tumores del SNC infantil son particularmente vulnerables.[49,117]
La evidencia de una adaptación psicosocial adversa después del cáncer infantil se ha obtenido de una variedad de fuentes que abarcan desde desenlaces notificados por pacientes o sus representantes hasta datos de registros poblacionales. La evidencia obtenida del primer tipo de fuente a veces es limitada por el tamaño pequeño de la muestra, el sesgo de selección de la cohorte y el sesgo de participación, así como por la variación de métodos y lugares (encuestas clínicas vs. encuestas a distancia) de las evaluaciones. La evidencia obtenida del segundo tipo de fuente a menudo no se correlaciona bien con las características clínicas y de tratamiento para permitir la identificación de sobrevivientes con riesgo alto de alteraciones psicosociales.
Idoneidad social
Los sobrevivientes con deficiencias neurocognitivas son particularmente vulnerables a presentar carencias en el logro de la idoneidad social esperada en la edad adulta.
Los sobrevivientes adultos de cáncer infantil que participaron en el estudio del CCSS y recibieron tratamiento con terapias dirigidas al SNC no lograron la independencia en tasas comparables a las de los sobrevivientes que no recibieron terapias dirigidas al SNC o sus homólogos hermanos. Se examinaron las relaciones entre los efectos tardíos neurológicos y el logro de la independencia en adultos sobrevivientes de cáncer tratados con terapias dirigidas al SNC (n = 7881) y sin estas (n = 8039) y una cohorte de hermanos (n = 3994). En comparación con los otros grupos, los sobrevivientes de cáncer infantil que recibieron tratamiento con terapias dirigidas al SNC mostraron tres patrones distintos de independencia funcional: 1) moderadamente independientes, nunca casados y vida no independiente (78,7 %); 2) moderadamente independientes, incapaces de conducir (15,6 %); 3) no independientes (5,7 %). Ningún sobreviviente de los que recibieron tratamiento con terapias dirigidas al SNC era totalmente independiente (una cuarta clase identificada). Por el contrario, el 50 % de los sobrevivientes que no recibieron tratamiento en el SNC y el 60 % de los hermanos se clasificaron como totalmente independientes. La no independencia se relacionó con síntomas de angustia emocional (es decir, depresión, ansiedad, somatización o ideas de suicidio).[118]
En un estudio poblacional de adultos sobrevivientes de tumores del SNC diagnosticados en la niñez o la adolescencia, los sobrevivientes tuvieron desenlaces significativamente más precarios en las funciones de autopercepción y autoestima que la población general. El sexo femenino, las secuelas físicas permanente y visibles, el tipo específico de tumor y el tratamiento con radioterapia craneal fueron factores de predicción de desenlaces precarios en la autopercepción.[119]
En una serie de sobrevivientes de neoplasias malignas del SNC (n = 802) notificada por el CCSS, los múltiples indicadores de resultados adversos de adaptación adulta exitosa (logro educativo, ingresos, empleo y estado civil) fueron más prevalentes en los sobrevivientes que notificaron una disfunción neurocognitiva.[42]
En conjunto, en los estudios en los que se evalúan los desenlaces psicosociales de los sobrevivientes de tumores del SNC se indican deficiencias en la competencia social que empeoran con el tiempo.[120] Estos incluyen problemas por el rechazo de pares y el aislamiento durante la niñez o la adolescencia, así como la incapacidad para cultivar amistades y mantener relaciones románticas como adultos.
En un análisis del estudio SJLIFE con 224 sobrevivientes de tumores del SNC (mediana de edad actual, 26 años; mediana de tiempo desde el diagnóstico, 18 años), el deterioro neurocognitivo se relacionó de forma significativa con logros educativos inferiores, desempleo y dependencia.[3]
En un subgrupo, las deficiencias significativas en las habilidades de percepción social, incluso el reconocimiento del afecto, contribuyeron al logro limitado de resultados funcionales. Los autoinformes de ajuste social se encontraban en general dentro de los límites normales, lo que indica una autopercepción limitada de las deficiencias sociales.[121]
En una serie con 1560 sobrevivientes adolescentes de LLA infantil tratados con quimioterapia sola, en el CCSS se identificó una proporción significativa de sobrevivientes que todavía experimentaban problemas de comportamiento obstinado, desatención e hiperactividad y aislamiento social, que se relacionaron con un aumento de riesgo de necesitar educación especial y pronosticaron logros educacionales reducidos en la edad adulta.[70]
En un estudio transversal de 855 sobrevivientes de leucemia infantil (media, 10,2 años desde el diagnóstico), los investigadores de la cohorte del Leucémie de l’Enfant et de l’Adolescent (LEA) identificaron factores independientes relacionados con la repetición de un grado escolar, como un bajo nivel educativo de los padres y problemas financieros en el hogar, resaltando la importancia de tener en cuenta los factores socioeconómicos que a veces afectan el acceso al apoyo educativo. Los sobrevivientes que eran adolescentes (edad 11–17 años) en el momento del diagnóstico también presentaron un riesgo mayor al de los sobrevivientes que eran niños (edad <11 años) en el momento del diagnóstico.[122]
En un estudio poblacional de supervivencia transversal, el 28 % de los sobrevivientes de sarcoma diagnosticado durante la adolescencia y adultez temprana (18–39 años en el momento del diagnóstico; mediana de tiempo desde el diagnóstico, 6,2 años) experimentaron deterioro del funcionamiento social.[123] En el análisis multivariante se identificaron relaciones independientes con deterioro del funcionamiento social y el desempleo (OR, 3,72; IC 95 %, 1,26–10,97) y la necesidad de hacer cambios en el estilo de vida debido a problemas financieros causados por el estado físico o el tratamiento médico de una persona (OR, 3,39; IC 95 %, 1,12–10,30). Por el contrario, un mejor apoyo social redujo el riesgo de deterioro del funcionamiento social (OR, 0,74; IC 95 %, 0,57–0,96).
Sufrimiento psicológico y tendencias suicidas
Los sobrevivientes de cáncer infantil también tienen riesgo de presentar síntomas de sufrimiento psicológico y tendencias suicidas.[124]
En un informe del CCSS se caracterizó la prevalencia y los factores de riesgo del malestar emocional autonotificado y la CVRS en sobrevivientes de rabdomiosarcoma (n = 713) y sus hermanos (n = 706). También se identificaron los factores de riesgo relacionados con estos desenlaces en los sobrevivientes.[78]
En comparación con sus hermanos, más sobrevivientes notificaron un malestar emocional elevado (malestar somático: 12,9 vs. 4,7 %; ansiedad: 11,7 vs. 5,9 %; depresión: 22,8 vs. 16,9 %) y una CVRS más precaria (funcionamiento físico: 11,1 vs. 2,8 %; desempeño en las actividades cotidianas debido a problemas físicos: 16,8 vs. 8,2 %; dolor: 17,5 vs. 10 %; vitalidad: 22,3 vs. 13,8 %; funcionamiento social: 14,4 vs. 6,8 %; funcionamiento emocional: 17,1 vs. 10,6 %).
La exposición a la radiación torácica y pélvica predijo un mayor riesgo de deterioro del funcionamiento físico (OR, 2,68 y 3,44, respectivamente).
El consumo de tabaco se relacionó con la ansiedad (OR, 2,71) y la depresión (OR, 1,77).
Las afecciones neurológicas aumentaron el riesgo de ansiedad (OR, 2,30) y las afecciones auditivas incrementaron el riesgo de depresión (OR, 1,79).
Se usó información del estudio SJLIFE para comparar el riesgo de ideación suicida, comportamiento suicida y mortalidad en adultos sobrevivientes de cáncer infantil en comparación con personas de la población general.[125]
Los sobrevivientes (n = 3096) notificaron una prevalencia a 12 meses de ideación suicida similar a la de la población general (razón de incidencia estandarizada [RIE], 0,68) y una prevalencia inferior de comportamiento suicida (planeación: CIE, 0,17; intentos: CIE, 0,07) y mortalidad (razón de mortalidad estandarizada, 0,60).
Entre los sobrevivientes, la depresión (RR, 12,30), la ansiedad (RR, 2,19), y el estrés financiero (RR, 1,47) fueron factores de riesgo relacionados con la ideación suicida.
Los sobrevivientes solteros, viudos o divorciados; quienes no trabajaban a tiempo completo y quienes notificaron estrés financiero durante el año previo o alteraciones del sueño presentaron una prevalencia de ideación suicida entre un 30 % a un 50 % más alta.
En un estudio del CCSS se evaluó la prevalencia de ideación suicida recidivante entre 9128 adultos sobrevivientes a largo plazo de cáncer infantil.[126]
Fue más probable que los sobrevivientes notificaran una ideación suicida tardía (OR, 1,9; IC 95 %, 1,5–2,5) e ideación suicida recurrente (OR, 2,6; IC 95 %, 1,8–3,8) en comparación con los hermanos.
Los antecedentes de convulsiones de los sobrevivientes se relacionaron con el doble de probabilidad de presentar ideación suicida.
En un estudio poblacional se evaluó el suicidio en adultos tratados por cáncer antes de los 25 años de edad.[127]
El riesgo absoluto de suicido fue bajo (24 casos entre 3375 muertes).
El CRI de suicidio aumentó en las personas tratadas por cáncer durante la niñez (0–14 años; CRI, 2,5; IC 95 %, 1,7–3,8) la adolescencia y adultez temprana (15–24 años; CRI, 2,3; IC 95 %, 1,2–4,6).
Las afecciones crónicas también pueden afectar aspectos de la salud psicológica.
En un estudio, en el que se evaluaron los desenlaces psicológicos en sobrevivientes a largo plazo tratados con TCMH, el 22 % de los sobrevivientes y el 8 % de los hermanos del grupo de control notificaron resultados adversos. El sufrimiento somático fue la afección más prevalente y afectó al 15 % de los sobrevivientes de TCMH, lo que representa un riesgo 3 veces más alto que el riesgo de los hermanos. Los sobrevivientes de TCMH con afecciones graves o potencialmente mortales y EICH activa crónica tuvieron un riesgo doble de sufrimiento somático.[128]
En un informe del CCSS, se notificó que la presencia de afecciones pulmonares, endocrinas y cardíacas crónicas en una muestra de 5021 adultos sobrevivientes de cáncer infantil se vinculó con un aumento del riesgo de síntomas de sufrimiento psicológico.[129]
En una investigación del CCSS, en la que se evaluaron los desenlaces psicológicos y educativos a largo plazo en sobrevivientes de neuroblastoma, los sobrevivientes tenían riesgo elevado de alteraciones psicológicas que se relacionaron con el uso de servicios de educación especial y menores logros académicos. La presencia de 2 o más afecciones crónicas, pero no las exposiciones terapéuticas comunes, fue predictiva de las alteraciones psicológicas. En concreto, la enfermedad pulmonar fue un factor de predicción de alteraciones en los 5 dominios psicológicos, mientras que la enfermedad endocrina y la neuropatía periférica fueron factores de predicción individual de alteraciones en 3 dominios.[130]
La incorporación de exámenes de detección psicológicos en las consultas médicas de los sobrevivientes de cáncer infantil puede ser valiosa. Sin embargo, la limitación de tales evaluaciones a quienes vuelven a los consultorios de seguimiento a largo plazo quizás produzca una submuestra sesgada de los sobrevivientes con más dificultades y sea más difícil establecer tasas precisas de prevalencia.
En un estudio de 101 adultos sobrevivientes de cáncer infantil, se realizaron exámenes de detección del ámbito psicológico durante una evaluación anual de rutina en el consultorio de supervivencia del Dana Farber Cancer Institute.[131]
En la Symptom Checklist 90 Revised, 32 sobrevivientes obtuvieron un resultado positivo (indicador de sufrimiento psicológico) y 14 sobrevivientes notificaron por lo menos un síntoma suicida.
Los factores de riesgo de sufrimiento psicológico fueron, entre otros, la insatisfacción de los sobrevivientes con su apariencia física, la salud física precaria y el tratamiento con irradiación craneal.
En este estudio, el instrumento demostró ser útil en el entorno de una visita al consultorio porque la evaluación sistemática de los aspectos psicológicos se completó en menos de 30 minutos. Además, en 80 % de los casos, no se generó malestar en los sobrevivientes al completar el instrumento.
En un estudio poblacional realizado en Taiwán se comparó la prevalencia de enfermedades mentales graves en 5121 sobrevivientes de cáncer diagnosticado durante la niñez o adolescencia con la de controles poblacionales.[132]
Los sobrevivientes (media de edad, 9 años; 60 % tenían 5 años de edad o más después del diagnóstico) mostraron un aumento del riesgo de 6 enfermedades mentales graves, en comparación con los controles. Estos trastornos incluyeron los siguientes:
Trastorno del espectro autista (CRI, 10,42; IC 95 %, 4,58–23,69).
Trastorno por déficit de atención o hiperactividad (CRI, 6,59; IC 95 %, 4,91–8,86).
Trastorno bipolar (CRI, 2,93; IC 95 %, 1,26–6,80).
Trastorno depresivo grave (CRI, 1,88; IC 95 %, 1,26–2,79).
Trastorno obsesivo compulsivo (TOC) (CRI, 3,37; IC 95 %, 1,33–8,52).
Trastorno de estrés postraumático (TEPT) (CRI, 6,10; IC 95 %, 1,46–25,54).
Los sobrevivientes eran más jóvenes que los controles en el momento del diagnóstico de TDAH, esquizofrenia, trastorno depresivo grave y TOC.
Los riesgos de enfermedades mentales graves variaron según los tipos de cáncer específicos. El mayor número de trastornos psiquiátricos mayores se observó entre los sobrevivientes de cáncer de encéfalo y cáncer de tejido linfático o hematopoyético.
En un estudio poblacional se vinculó a personas con antecedentes de 6 cánceres comunes diagnosticados entre los 15 y los 21 años de edad con datos provinciales de atención sanitaria para comparar las tasas de visitas ambulatorias (médico de cabecera y psiquiatra) por indicaciones psiquiátricas y el tiempo transcurrido hasta que se produjeron eventos psiquiátricos graves (visitas a urgencias, hospitalización y suicidio). El estudio incluyó a 2208 pacientes de cáncer diagnosticado durante la adolescencia y adultez temprana y a 10 457 controles emparejados.[133]
Los sobrevivientes a 5 años de un cáncer diagnosticado durante la adolescencia o adultez temprana experimentaron mayores tasas de visitas ambulatorias de salud mental que los controles (671 frente a 506 visitas por 1000 personas-año; RR, 1,3), mayor riesgo de episodio psiquiátrico grave (CRI, 1,2) y mayor riesgo de episodio grave relacionado con un trastorno psicótico (CRI, 2,0).
El tratamiento en un centro de adultos se relacionó con tasas de visitas ambulatorias sustancialmente mayores, en comparación con el tratamiento en centros pediátricos (RR, 1,8).
Trastorno de estrés postraumático después del cáncer infantil
A pesar del exceso de factores estresantes relacionados con el diagnóstico de cáncer y su tratamiento, en general, en los estudios se revelan índices bajos de síntomas de estrés postraumático y trastorno por estrés postraumático (TEPT) en niños con cáncer, que por lo habitual no superan los índices en los niños sanos en las comparaciones.[134]
Se notificó que la prevalencia de TEPT y síntomas de estrés postraumático es del 15 % al 20 % en los adultos jóvenes sobrevivientes de cáncer infantil, con cálculos variables según los criterios utilizados para definir estas afecciones.[135]
En una cohorte de 5121 sobrevivientes de cáncer diagnosticado durante la niñez y adolescencia (media de edad, 9 años), la incidencia de TEPT fue del 0,59 %. En comparación con los controles, los sobrevivientes presentaron un riesgo 6,10 veces más alto de recibir un diagnóstico de TEPT. Los sobrevivientes de cáncer de encéfalo tuvieron el riesgo más alto de todos los diagnósticos de cáncer de TEPT (CRI, 18,50; IC 95 %, 2,11–162,64).[132]
El estilo de adaptación del paciente y sus progenitores son factores determinantes importantes de TEPT en el entorno de la oncología pediátrica.[136,137]
Los sobrevivientes con TEPT notificaron más problemas psicológicos y creencias negativas acerca de su enfermedad y estado de salud que aquellos sin este trastorno.[138,139]
Un subconjunto de adultos sobrevivientes (9 %) que participaron en el CCSS notificó deterioro funcional o sufrimiento clínico además del conjunto de síntomas compatibles con un diagnóstico completo de TEPT.[140]
El TEPT fue significativamente más frecuente en los sobrevivientes que en las comparaciones con los hermanos.
El TEPT se relacionó estrechamente con ser soltero, tener ingresos anuales inferiores a $20 000, estar desempleado, contar con estudios secundarios o inferiores y ser mayor de 30 años.
En particular, los sobrevivientes tratados con irradiación craneal antes de los 4 años tuvieron un riesgo alto de presentar TEPT.
El tratamiento intensivo del cáncer también se relacionó con un aumento del riesgo de TEPT pleno.
Dado que evitar lugares y personas relacionadas con el cáncer forma parte del TEPT, el síndrome a veces interfiere con la obtención de atención médica adecuada.[141]
Aquellas personas con TEPT perciben mayores amenazas actuales para su vida o la vida de sus hijos.
Otros factores de riesgo de TEPT son el funcionamiento familiar deficiente, la disminución del apoyo social y los factores de estrés que no se relacionan con el cáncer.
Consecuencias psicosociales en sobrevivientes de cáncer diagnosticado durante la niñez, adolescencia y adultez temprana
La mayor parte de la investigación sobre los efectos tardíos después del cáncer se ha centrado en personas que presentaron cáncer diagnosticado durante la niñez. Se sabe poco acerca del efecto específico de un diagnóstico de cáncer durante la adolescencia o del efecto del cáncer infantil en los desenlaces psicosociales de los adolescentes y adultos jóvenes (AAJ).
Evidencia (desenlaces psicosociales en sobrevivientes de cáncer diagnosticado durante la adolescencia o adultez temprana):
Se comparó un grupo de adultos sobrevivientes de cáncer diagnosticado durante la adolescencia (entre los 15 y 18 años) (n = 825) con una muestra ajustada por edad de la población general y un grupo de comparación de adultos sin cáncer.[142]
Las mujeres sobrevivientes de cáncer diagnosticado durante la adolescencia alcanzaron menos hitos del desarrollo psicosexual, como tener su primer novio, o lo hicieron más tarde.
Fue más probable que los varones sobrevivientes vivieran con sus padres que los controles del mismo sexo.
Fue menos probable que los sobrevivientes de cáncer diagnosticado durante la adolescencia se hubieran casado o tenido hijos. Los sobrevivientes tenían muchos más años en el momento de casarse por primera vez y en el momento del nacimiento de su primer hijo que aquellos controles de la muestra ajustada por edad.
Los sobrevivientes de esta cohorte estaban mucho menos satisfechos con su vida y su salud en términos generales que las personas de un grupo de control de la comunidad. El deterioro de la satisfacción en general y la relacionada con la salud se vinculó con efectos tardíos somáticos, síntomas de depresión y ansiedad, y tasas más bajas de crecimiento postraumático.[143]
En una encuesta de 4054 sobrevivientes de cáncer diagnosticado durante la adolescencia o adultez temprana y 345 592 entrevistados sin antecedentes de esta enfermedad, se notificaron los siguientes resultados:[144]
Los sobrevivientes de cáncer diagnosticado durante la adolescencia o adultez temprana eran más propensos a fumar (26 vs. 18 %), presentar obesidad (31 vs. 9 %) y sufrir de afecciones crónicas como enfermedad cardiovascular (14 vs. 7 %), hipertensión (35 vs. 9 %), asma (15 vs. 8 %), incapacidades (36 vs 18 %) y salud mental precaria (20 vs. 10 %).
También fue menos probable que recibieran atención médica debido al costo (24 vs. 15 %).
En el CCSS se evaluaron los desenlaces en 2979 sobrevivientes de cáncer diagnosticado durante la adolescencia y 649 hermanos de ambos sexos de sobrevivientes de cáncer para determinar la incidencia de dificultades en 6 dominios conductuales y sociales (depresión o ansiedad, comportamiento obstinado, déficit de atención, conflicto con los pares o aislamiento social, comportamientos antisociales y aptitudes sociales).[145]
Los sobrevivientes tuvieron una probabilidad 1,5 veces más alta (IC 95 %, 1,1–2,1) que los hermanos de presentar síntomas de depresión o ansiedad, y 1,7 veces más alta de exhibir comportamientos antisociales que los hermanos (IC 95 %, 1,3–2,2).
Las puntuaciones en los dominios de depresión o ansiedad, déficit de atención y comportamiento antisocial fueron significativamente elevadas en los adolescentes tratados por leucemia o tumores del SNC en comparación con las puntuaciones de los hermanos.
Además, los sobrevivientes de neuroblastoma tuvieron dificultades en los dominios de depresión o ansiedad y comportamiento antisocial.
Los tratamientos dirigidos al SNC (radioterapia craneal o metotrexato intratecal) fueron factores de riesgo específicos de desenlaces conductuales adversos.
En otro estudio del CCSS, se evaluó el funcionamiento psicológico y neurocognitivo de 2589 sobrevivientes a largo plazo de un cáncer diagnosticado cuando eran adolescentes o adultos jóvenes.[146]
En comparación con la cohorte de hermanos, los sobrevivientes que recibieron el diagnóstico siendo adolescentes o adultos jóvenes notificaron tasas más altas de depresión (OR, 1,55; IC 95 %, 1,04–2,30) y ansiedad (OR, 2,00; IC 95 %, 1,17–3,43) y más problemas cognitivos que afectaban el eficiencia de tareas (OR, 1,72; IC 95 %, 1,21–2,43), la regulación emocional (OR, 1,74; IC 95 %, 1,26–2,40) y la memoria (OR, 1,44; IC 95 %, 1,09–1,89).
Los sobrevivientes de linfoma y sarcoma que recibieron el diagnóstico durante la adolescencia tardía tuvieron menor riesgo de problemas psicosociales y neurocognitivos en comparación con quienes recibieron el diagnóstico antes de los 11 años de edad. Estos desenlaces no difirieron según la edad en el momento del diagnóstico en los sobrevivientes de leucemia o tumores del SNC.
Fue menos probable que aquellos sobrevivientes que recibieron el diagnóstico cuando eran adolescentes o adultos jóvenes obtuvieran una educación universitaria, trabajaran a tiempo completo, contrajeran matrimonio o vivieran de forma independiente en comparación con sus hermanos del grupo de control; los desenlaces inferiores en el dominio social se relacionaron con síntomas neurocognitivos.
En un estudio de seguimiento del CCSS se evaluaron los perfiles de síntomas comórbidos de 3993 adolescentes (13–17 años) tratados por cáncer.[147] En el análisis del perfil latente se identificaron los siguientes 4 perfiles de síntomas:
Sin síntomas importantes.
Síntomas internalizados elevados (ansiedad o depresión, retraimiento social y problemas de atención).
Síntomas externalizados elevados (comportamiento obstinado y problemas de atención).
Síntomas internalizados y externalizados elevados.
Los resultados generales apoyan la idea de que los síntomas conductuales, emocionales y sociales se suelen presentar en forma simultánea y se vinculan con exposiciones al tratamiento (radiación craneal, corticoesteroides y metotrexato) y efectos tardíos (obesidad, dolor relacionado con el cáncer y alteraciones sensoriales) en los adolescentes sobrevivientes que se diagnosticaron entre 1970 y 1986.
En otro estudio del CCSS se caracterizó la prevalencia del riesgo de dolor, la interferencia clínicamente significativa del dolor en las actividades diarias y el dolor recurrente en 10 012 adultos sobrevivientes de cáncer infantil (mediana de tiempo desde el diagnóstico, 23 años).[148]
Una minoría significativa de sobrevivientes refirió dolor de moderado a grave (29 %), interferencia del dolor de moderada a extrema (20 %) y dolor recurrente de moderado a grave (9 %).
La edad avanzada en el momento del diagnóstico y el seguimiento, el sexo femenino y la presencia de afecciones crónicas de grados 3 a 4 se asociaron con un aumento del riesgo de peores desenlaces respecto al dolor.
Pertenecer a grupos de minorías raciales o étnicas, tener un diagnóstico de tumor del SNC y el tratamiento de quimioterapia con derivados de platino y radiación craneal se relacionaron con un aumento del riesgo de dolor de presentación tardía e interferencia del dolor.
La depresión y la ansiedad se vincularon con un aumento del riesgo de todos los desenlaces de dolor, y la vitalidad precaria intervino en los efectos de la ansiedad sobre el dolor intenso y la interferencia del dolor.
En un análisis del estudio SJLIFE se evaluaron las asociaciones entre las evaluaciones médicas, neurocognitivas y físicas detallada, además del dolor autonotificado, la calidad de vida y el funcionamiento social en 2836 sobrevivientes (media de edad, 32,2 años; media de tiempo desde el diagnóstico, 23,7 años) y 343 controles comunitarios sin cáncer.[149]
El dolor moderado a muy intenso que interfiere de manera moderada a extrema con las actividades diarias fue notificado por el 18 % de los sobrevivientes, en comparación con el 8 % de los controles (P < 0,001).
Los sobrevivientes de sarcoma de tejido blando (OR, 9,25), linfoma no Hodgkin (OR, 4,13) y sarcoma de Ewing u osteosarcoma (OR, 3,93) tenían la probabilidad más alta de dolor con interferencia diaria, en comparación con los controles.
En modelos multivariantes, las exposiciones a tratamientos, los antecedentes de amputación (OR, 1,89) y la cirugía con conservación del miembro (OR, 2,30) fueron las características asociadas con aumento de la probabilidad de interferencia diaria.
El dolor con interferencia diaria se asoció con aumento del riesgo de deterioro neurocognitivo (atención: RR, 1,88; memoria: RR, 1,65) y funcionamiento físico (capacidad aeróbica: RR, 2,29; movilidad: RR, 1,71).
El dolor con interferencia diaria también se asoció con aumento del riesgo de deterioro del funcionamiento social (incapacidad de mantenerse empleado o asistir a la escuela: RR, 4,46; ayuda con las necesidades rutinarias o de cuidado personal: RR, 5,64), y calidad de vida relacionada con la salud (física: RR, 6,34; emocional: RR, 2,83).
En un estudio de cohorte de registros nórdico, se evaluó si los sobrevivientes de cáncer infantil (n = 18 621) tenían un riesgo más alto de presentar trastornos psiquiátricos más adelante en la vida, en comparación con sus hermanos (n = 24 775) y la población general (n = 88 630).[150]
La incidencia acumulada de contacto con un hospital psiquiátrico a la edad de 30 años fue del 15,9 % en los sobrevivientes de cáncer infantil, del 14,0 % en los hermanos y del 12,7 % en los controles poblacionales emparejados (por año de nacimiento, país o municipio).
La diferencia absoluta fue pequeña, pero los sobrevivientes tenían un RR más alto de un contacto con un hospital psiquiátrico que sus hermanos (1,39) y los controles emparejados (CRI, 1,34).
En un estudio del CCSS se evaluaron las relaciones entre los cambios de tratamiento y las capacidades neurocognitivas, así como el efecto de las capacidades neurocognitivas y las afecciones crónicas sobre la independencia en la edad adulta en 1284 sobrevivientes adultos de gliomas infantiles (mediana de edad, 30 años en el momento de la evaluación y 22 años desde el diagnóstico).[151]
La exposición a la radiación craneal disminuyó durante el período de estudio (década de 1970–década de 1990).
Los sobrevivientes con cualquier exposición al tratamiento (cirugía sola, quimioterapia con cirugía o sin esta, radioterapia craneal con quimioterapia o cirugía o sin estas) tenían un riesgo elevado de deterioro neurocognitivo, en comparación con los hermanos.
Aunque la mayoría de los sobrevivientes de glioma a largo plazo de la cohorte alcanzaron la independencia en la edad adulta, se observaron relaciones con el deterioro neurocognitivo asociado al tratamiento y con afecciones crónicas entre aquellos sin independencia funcional.
Evidencia (independencia funcional y social):
En un estudio de 665 sobrevivientes de tumores del SNC (54 % varones; 52 % tratados con radioterapia craneal; mediana de edad, 15 años; y 12 años desde el diagnóstico), los investigadores del CCSS observaron los siguientes resultados:[117]
Casi el 50 % de los sobrevivientes presentaron dificultades sociales en las relaciones con sus pares que excedían las dificultades de los sobrevivientes de tumores sólidos y de los hermanos del grupo de control.
La exposición a radiación craneal predijo la dificultad en las relaciones sociales y de pares, además las alteraciones cognitivas intervinieron en estas asociaciones.
En un análisis del estudio SJLIFE se investigó la independencia funcional y social en 306 sobrevivientes de tumores del SNC (astrocitoma [n = 130], meduloblastoma [n = 77], ependimoma [n = 36], y otros [n = 63]; mediana de edad, 25 años; y tiempo desde el diagnóstico, 16,8 años).[49]
Solo el 40 % de los sobrevivientes a largo plazo en esta cohorte lograron la independencia completa como adultos.
Los factores pronósticos de la falta de independencia fueron el tratamiento con irradiación craneoespinal, antecedentes de hidrocefalia con derivación y una edad más joven en el momento del diagnóstico.
Además de la alteración en la puntuación de CI, las limitaciones funcionales en la capacidad aeróbica, la flexibilidad y el funcionamiento físico adaptativo se relacionaron de manera significativa con la falta de independencia.
Bibliografía
Grill J, Kieffer V, Kalifa C: Measuring the neuro-cognitive side-effects of irradiation in children with brain tumors. Pediatr Blood Cancer 42 (5): 452-6, 2004. [PUBMED Abstract]
Marusak HA, Iadipaolo AS, Harper FW, et al.: Neurodevelopmental consequences of pediatric cancer and its treatment: applying an early adversity framework to understanding cognitive, behavioral, and emotional outcomes. Neuropsychol Rev 28 (2): 123-175, 2018. [PUBMED Abstract]
Brinkman TM, Krasin MJ, Liu W, et al.: Long-Term Neurocognitive Functioning and Social Attainment in Adult Survivors of Pediatric CNS Tumors: Results From the St Jude Lifetime Cohort Study. J Clin Oncol 34 (12): 1358-67, 2016. [PUBMED Abstract]
Moxon-Emre I, Bouffet E, Taylor MD, et al.: Impact of craniospinal dose, boost volume, and neurologic complications on intellectual outcome in patients with medulloblastoma. J Clin Oncol 32 (17): 1760-8, 2014. [PUBMED Abstract]
Krull KR, Zhang N, Santucci A, et al.: Long-term decline in intelligence among adult survivors of childhood acute lymphoblastic leukemia treated with cranial radiation. Blood 122 (4): 550-3, 2013. [PUBMED Abstract]
Annett RD, Hile S, Bedrick E, et al.: Neuropsychological functioning of children treated for acute lymphoblastic leukemia: impact of whole brain radiation therapy. Psychooncology 24 (2): 181-9, 2015. [PUBMED Abstract]
van der Plas E, Qiu W, Nieman BJ, et al.: Sex-Specific Associations Between Chemotherapy, Chronic Conditions, and Neurocognitive Impairment in Acute Lymphoblastic Leukemia Survivors: A Report From the Childhood Cancer Survivor Study. J Natl Cancer Inst 113 (5): 588-596, 2021. [PUBMED Abstract]
Krull KR, Khan RB, Ness KK, et al.: Symptoms of attention-deficit/hyperactivity disorder in long-term survivors of childhood leukemia. Pediatr Blood Cancer 57 (7): 1191-6, 2011. [PUBMED Abstract]
Kahalley LS, Conklin HM, Tyc VL, et al.: ADHD and secondary ADHD criteria fail to identify many at-risk survivors of pediatric ALL and brain tumor. Pediatr Blood Cancer 57 (1): 110-8, 2011. [PUBMED Abstract]
Mahajan A, Stavinoha PL, Rongthong W, et al.: Neurocognitive Effects and Necrosis in Childhood Cancer Survivors Treated With Radiation Therapy: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 401-416, 2024. [PUBMED Abstract]
Cheung YT, Brinkman TM, Li C, et al.: Chronic Health Conditions and Neurocognitive Function in Aging Survivors of Childhood Cancer: A Report from the Childhood Cancer Survivor Study. J Natl Cancer Inst 110 (4): 411-419, 2018. [PUBMED Abstract]
Bass JK, Liu W, Banerjee P, et al.: Association of Hearing Impairment With Neurocognition in Survivors of Childhood Cancer. JAMA Oncol 6 (9): 1363-1371, 2020. [PUBMED Abstract]
Barlow-Krelina E, Chen Y, Yasui Y, et al.: Consistent Physical Activity and Future Neurocognitive Problems in Adult Survivors of Childhood Cancers: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 38 (18): 2041-2052, 2020. [PUBMED Abstract]
Bernal JDK, Recchia F, Yu DJ, et al.: Physical activity and exercise for cancer-related cognitive impairment among individuals affected by childhood cancer: a systematic review and meta-analysis. Lancet Child Adolesc Health 7 (1): 47-58, 2023. [PUBMED Abstract]
Phillips NS, Stratton KL, Williams AM, et al.: Late-onset Cognitive Impairment and Modifiable Risk Factors in Adult Childhood Cancer Survivors. JAMA Netw Open 6 (5): e2316077, 2023. [PUBMED Abstract]
Traunwieser T, Loos E, Ottensmeier H, et al.: Survivors of infant atypical teratoid/rhabdoid tumors present with severely impaired cognitive functions especially for fluid intelligence and visual processing: data from the German brain tumor studies. Pediatr Blood Cancer 71 (5): e30910, 2024. [PUBMED Abstract]
Merchant TE, Schreiber JE, Wu S, et al.: Critical combinations of radiation dose and volume predict intelligence quotient and academic achievement scores after craniospinal irradiation in children with medulloblastoma. Int J Radiat Oncol Biol Phys 90 (3): 554-61, 2014. [PUBMED Abstract]
Armstrong GT, Conklin HM, Huang S, et al.: Survival and long-term health and cognitive outcomes after low-grade glioma. Neuro Oncol 13 (2): 223-34, 2011. [PUBMED Abstract]
Di Pinto M, Conklin HM, Li C, et al.: Learning and memory following conformal radiation therapy for pediatric craniopharyngioma and low-grade glioma. Int J Radiat Oncol Biol Phys 84 (3): e363-9, 2012. [PUBMED Abstract]
Ris MD, Walsh K, Wallace D, et al.: Intellectual and academic outcome following two chemotherapy regimens and radiotherapy for average-risk medulloblastoma: COG A9961. Pediatr Blood Cancer 60 (8): 1350-7, 2013. [PUBMED Abstract]
Bowers DC, Liu Y, Leisenring W, et al.: Late-occurring stroke among long-term survivors of childhood leukemia and brain tumors: a report from the Childhood Cancer Survivor Study. J Clin Oncol 24 (33): 5277-82, 2006. [PUBMED Abstract]
Nassar SL, Conklin HM, Zhou Y, et al.: Neurocognitive outcomes among children who experienced seizures during treatment for acute lymphoblastic leukemia. Pediatr Blood Cancer 64 (8): , 2017. [PUBMED Abstract]
Torres VA, Ashford JM, Wright E, et al.: The impact of socioeconomic status (SES) on cognitive outcomes following radiotherapy for pediatric brain tumors: a prospective, longitudinal trial. Neuro Oncol 23 (7): 1173-1182, 2021. [PUBMED Abstract]
Mabbott DJ, Spiegler BJ, Greenberg ML, et al.: Serial evaluation of academic and behavioral outcome after treatment with cranial radiation in childhood. J Clin Oncol 23 (10): 2256-63, 2005. [PUBMED Abstract]
Brière ME, Scott JG, McNall-Knapp RY, et al.: Cognitive outcome in pediatric brain tumor survivors: delayed attention deficit at long-term follow-up. Pediatr Blood Cancer 50 (2): 337-40, 2008. [PUBMED Abstract]
Edelstein K, Spiegler BJ, Fung S, et al.: Early aging in adult survivors of childhood medulloblastoma: long-term neurocognitive, functional, and physical outcomes. Neuro Oncol 13 (5): 536-45, 2011. [PUBMED Abstract]
Palmer SL, Goloubeva O, Reddick WE, et al.: Patterns of intellectual development among survivors of pediatric medulloblastoma: a longitudinal analysis. J Clin Oncol 19 (8): 2302-8, 2001. [PUBMED Abstract]
Mulhern RK, Merchant TE, Gajjar A, et al.: Late neurocognitive sequelae in survivors of brain tumours in childhood. Lancet Oncol 5 (7): 399-408, 2004. [PUBMED Abstract]
Brinkman TM, Reddick WE, Luxton J, et al.: Cerebral white matter integrity and executive function in adult survivors of childhood medulloblastoma. Neuro Oncol 14 (Suppl 4): iv25-36, 2012. [PUBMED Abstract]
Jacola LM, Ashford JM, Reddick WE, et al.: The relationship between working memory and cerebral white matter volume in survivors of childhood brain tumors treated with conformal radiation therapy. J Neurooncol 119 (1): 197-205, 2014. [PUBMED Abstract]
Palmer SL, Glass JO, Li Y, et al.: White matter integrity is associated with cognitive processing in patients treated for a posterior fossa brain tumor. Neuro Oncol 14 (9): 1185-93, 2012. [PUBMED Abstract]
Ali JS, Ashford JM, Swain MA, et al.: Predictors of Cognitive Performance Among Infants Treated for Brain Tumors: Findings From a Multisite, Prospective, Longitudinal Trial. J Clin Oncol 39 (21): 2350-2358, 2021. [PUBMED Abstract]
Merchant TE, Conklin HM, Wu S, et al.: Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: prospective evaluation of cognitive, endocrine, and hearing deficits. J Clin Oncol 27 (22): 3691-7, 2009. [PUBMED Abstract]
Liu APY, Hastings C, Wu S, et al.: Treatment burden and long-term health deficits of patients with low-grade gliomas or glioneuronal tumors diagnosed during the first year of life. Cancer 125 (7): 1163-1175, 2019. [PUBMED Abstract]
Palmer SL, Armstrong C, Onar-Thomas A, et al.: Processing speed, attention, and working memory after treatment for medulloblastoma: an international, prospective, and longitudinal study. J Clin Oncol 31 (28): 3494-500, 2013. [PUBMED Abstract]
Knight SJ, Conklin HM, Palmer SL, et al.: Working memory abilities among children treated for medulloblastoma: parent report and child performance. J Pediatr Psychol 39 (5): 501-11, 2014. [PUBMED Abstract]
Schreiber JE, Gurney JG, Palmer SL, et al.: Examination of risk factors for intellectual and academic outcomes following treatment for pediatric medulloblastoma. Neuro Oncol 16 (8): 1129-36, 2014. [PUBMED Abstract]
Khan RB, Patay Z, Klimo P, et al.: Clinical features, neurologic recovery, and risk factors of postoperative posterior fossa syndrome and delayed recovery: a prospective study. Neuro Oncol 23 (9): 1586-1596, 2021. [PUBMED Abstract]
Schreiber JE, Palmer SL, Conklin HM, et al.: Posterior fossa syndrome and long-term neuropsychological outcomes among children treated for medulloblastoma on a multi-institutional, prospective study. Neuro Oncol 19 (12): 1673-1682, 2017. [PUBMED Abstract]
Moxon-Emre I, Taylor MD, Bouffet E, et al.: Intellectual Outcome in Molecular Subgroups of Medulloblastoma. J Clin Oncol 34 (34): 4161-4170, 2016. [PUBMED Abstract]
Acharya S, Wu S, Ashford JM, et al.: Association between hippocampal dose and memory in survivors of childhood or adolescent low-grade glioma: a 10-year neurocognitive longitudinal study. Neuro Oncol 21 (9): 1175-1183, 2019. [PUBMED Abstract]
Ellenberg L, Liu Q, Gioia G, et al.: Neurocognitive status in long-term survivors of childhood CNS malignancies: a report from the Childhood Cancer Survivor Study. Neuropsychology 23 (6): 705-17, 2009. [PUBMED Abstract]
Armstrong GT, Liu Q, Yasui Y, et al.: Long-term outcomes among adult survivors of childhood central nervous system malignancies in the Childhood Cancer Survivor Study. J Natl Cancer Inst 101 (13): 946-58, 2009. [PUBMED Abstract]
Ris MD, Leisenring WM, Goodman P, et al.: Neuropsychological and socioeconomic outcomes in adult survivors of pediatric low-grade glioma. Cancer 125 (17): 3050-3058, 2019. [PUBMED Abstract]
Shabason EK, Brodsky C, Baran J, et al.: Clinical diagnosis of attention-deficit/hyperactivity disorder in survivors of pediatric brain tumors. J Neurooncol 143 (2): 305-312, 2019. [PUBMED Abstract]
Brinkman TM, Palmer SL, Chen S, et al.: Parent-reported social outcomes after treatment for pediatric embryonal tumors: a prospective longitudinal study. J Clin Oncol 30 (33): 4134-40, 2012. [PUBMED Abstract]
Moyer KH, Willard VW, Gross AM, et al.: The impact of attention on social functioning in survivors of pediatric acute lymphoblastic leukemia and brain tumors. Pediatr Blood Cancer 59 (7): 1290-5, 2012. [PUBMED Abstract]
Mitby PA, Robison LL, Whitton JA, et al.: Utilization of special education services and educational attainment among long-term survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. Cancer 97 (4): 1115-26, 2003. [PUBMED Abstract]
Brinkman TM, Ness KK, Li Z, et al.: Attainment of Functional and Social Independence in Adult Survivors of Pediatric CNS Tumors: A Report From the St Jude Lifetime Cohort Study. J Clin Oncol 36 (27): 2762-2769, 2018. [PUBMED Abstract]
Kahalley LS, Ris MD, Grosshans DR, et al.: Comparing Intelligence Quotient Change After Treatment With Proton Versus Photon Radiation Therapy for Pediatric Brain Tumors. J Clin Oncol 34 (10): 1043-9, 2016. [PUBMED Abstract]
Pulsifer MB, Sethi RV, Kuhlthau KA, et al.: Early Cognitive Outcomes Following Proton Radiation in Pediatric Patients With Brain and Central Nervous System Tumors. Int J Radiat Oncol Biol Phys 93 (2): 400-7, 2015. [PUBMED Abstract]
Kahalley LS, Peterson R, Ris MD, et al.: Superior Intellectual Outcomes After Proton Radiotherapy Compared With Photon Radiotherapy for Pediatric Medulloblastoma. J Clin Oncol 38 (5): 454-461, 2020. [PUBMED Abstract]
Reddick WE, Glass JO, Helton KJ, et al.: Prevalence of leukoencephalopathy in children treated for acute lymphoblastic leukemia with high-dose methotrexate. AJNR Am J Neuroradiol 26 (5): 1263-9, 2005. [PUBMED Abstract]
Waber DP, Queally JT, Catania L, et al.: Neuropsychological outcomes of standard risk and high risk patients treated for acute lymphoblastic leukemia on Dana-Farber ALL consortium protocol 95-01 at 5 years post-diagnosis. Pediatr Blood Cancer 58 (5): 758-65, 2012. [PUBMED Abstract]
Krull KR, Brinkman TM, Li C, et al.: Neurocognitive outcomes decades after treatment for childhood acute lymphoblastic leukemia: a report from the St Jude lifetime cohort study. J Clin Oncol 31 (35): 4407-15, 2013. [PUBMED Abstract]
Reddick WE, Shan ZY, Glass JO, et al.: Smaller white-matter volumes are associated with larger deficits in attention and learning among long-term survivors of acute lymphoblastic leukemia. Cancer 106 (4): 941-9, 2006. [PUBMED Abstract]
Kadan-Lottick NS, Zeltzer LK, Liu Q, et al.: Neurocognitive functioning in adult survivors of childhood non-central nervous system cancers. J Natl Cancer Inst 102 (12): 881-93, 2010. [PUBMED Abstract]
Schuitema I, Deprez S, Van Hecke W, et al.: Accelerated aging, decreased white matter integrity, and associated neuropsychological dysfunction 25 years after pediatric lymphoid malignancies. J Clin Oncol 31 (27): 3378-88, 2013. [PUBMED Abstract]
Armstrong GT, Reddick WE, Petersen RC, et al.: Evaluation of memory impairment in aging adult survivors of childhood acute lymphoblastic leukemia treated with cranial radiotherapy. J Natl Cancer Inst 105 (12): 899-907, 2013. [PUBMED Abstract]
Spiegler BJ, Kennedy K, Maze R, et al.: Comparison of long-term neurocognitive outcomes in young children with acute lymphoblastic leukemia treated with cranial radiation or high-dose or very high-dose intravenous methotrexate. J Clin Oncol 24 (24): 3858-64, 2006. [PUBMED Abstract]
Campbell LK, Scaduto M, Sharp W, et al.: A meta-analysis of the neurocognitive sequelae of treatment for childhood acute lymphocytic leukemia. Pediatr Blood Cancer 49 (1): 65-73, 2007. [PUBMED Abstract]
Cheung YT, Sabin ND, Reddick WE, et al.: Leukoencephalopathy and long-term neurobehavioural, neurocognitive, and brain imaging outcomes in survivors of childhood acute lymphoblastic leukaemia treated with chemotherapy: a longitudinal analysis. Lancet Haematol 3 (10): e456-e466, 2016. [PUBMED Abstract]
Jansen NC, Kingma A, Schuitema A, et al.: Neuropsychological outcome in chemotherapy-only-treated children with acute lymphoblastic leukemia. J Clin Oncol 26 (18): 3025-30, 2008. [PUBMED Abstract]
Iyer NS, Balsamo LM, Bracken MB, et al.: Chemotherapy-only treatment effects on long-term neurocognitive functioning in childhood ALL survivors: a review and meta-analysis. Blood 126 (3): 346-53, 2015. [PUBMED Abstract]
Jacola LM, Krull KR, Pui CH, et al.: Longitudinal Assessment of Neurocognitive Outcomes in Survivors of Childhood Acute Lymphoblastic Leukemia Treated on a Contemporary Chemotherapy Protocol. J Clin Oncol 34 (11): 1239-47, 2016. [PUBMED Abstract]
Espy KA, Moore IM, Kaufmann PM, et al.: Chemotherapeutic CNS prophylaxis and neuropsychologic change in children with acute lymphoblastic leukemia: a prospective study. J Pediatr Psychol 26 (1): 1-9, 2001 Jan-Feb. [PUBMED Abstract]
Buizer AI, de Sonneville LM, Veerman AJ: Effects of chemotherapy on neurocognitive function in children with acute lymphoblastic leukemia: a critical review of the literature. Pediatr Blood Cancer 52 (4): 447-54, 2009. [PUBMED Abstract]
Gandy K, Sapkota Y, Scoggins MA, et al.: Genetic variants, neurocognitive outcomes, and functional neuroimaging in survivors of childhood acute lymphoblastic leukemia. JNCI Cancer Spectr 7 (4): , 2023. [PUBMED Abstract]
Cole PD, Finkelstein Y, Stevenson KE, et al.: Polymorphisms in Genes Related to Oxidative Stress Are Associated With Inferior Cognitive Function After Therapy for Childhood Acute Lymphoblastic Leukemia. J Clin Oncol 33 (19): 2205-11, 2015. [PUBMED Abstract]
Jacola LM, Edelstein K, Liu W, et al.: Cognitive, behaviour, and academic functioning in adolescent and young adult survivors of childhood acute lymphoblastic leukaemia: a report from the Childhood Cancer Survivor Study. Lancet Psychiatry 3 (10): 965-972, 2016. [PUBMED Abstract]
Jacola LM, Conklin HM, Krull KR, et al.: The Impact of Intensified CNS-Directed Therapy on Neurocognitive Outcomes in Survivors of Childhood Acute Lymphoblastic Leukemia Treated Without Cranial Irradiation. J Clin Oncol 40 (36): 4218-4227, 2022. [PUBMED Abstract]
Krull KR, Cheung YT, Liu W, et al.: Chemotherapy Pharmacodynamics and Neuroimaging and Neurocognitive Outcomes in Long-Term Survivors of Childhood Acute Lymphoblastic Leukemia. J Clin Oncol 34 (22): 2644-53, 2016. [PUBMED Abstract]
Halsey C, Buck G, Richards S, et al.: The impact of therapy for childhood acute lymphoblastic leukaemia on intelligence quotients; results of the risk-stratified randomized central nervous system treatment trial MRC UKALL XI. J Hematol Oncol 4: 42, 2011. [PUBMED Abstract]
Kadan-Lottick NS, Brouwers P, Breiger D, et al.: A comparison of neurocognitive functioning in children previously randomized to dexamethasone or prednisone in the treatment of childhood acute lymphoblastic leukemia. Blood 114 (9): 1746-52, 2009. [PUBMED Abstract]
Williams AM, Cheung YT, Hyun G, et al.: Childhood Neurotoxicity and Brain Resilience to Adverse Events during Adulthood. Ann Neurol 89 (3): 534-545, 2021. [PUBMED Abstract]
Stefanski KJ, Anixt JS, Goodman P, et al.: Long-Term Neurocognitive and Psychosocial Outcomes After Acute Myeloid Leukemia: A Childhood Cancer Survivor Study Report. J Natl Cancer Inst 113 (4): 481-495, 2021. [PUBMED Abstract]
Edelmann MN, Daryani VM, Bishop MW, et al.: Neurocognitive and Patient-Reported Outcomes in Adult Survivors of Childhood Osteosarcoma. JAMA Oncol 2 (2): 201-8, 2016. [PUBMED Abstract]
van der Plas E, Darji H, Srivastava DK, et al.: Risk factors for neurocognitive impairment, emotional distress, and poor quality of life in survivors of pediatric rhabdomyosarcoma: A report from the Childhood Cancer Survivor Study. Cancer 130 (12): 2224-2236, 2024. [PUBMED Abstract]
Tonning Olsson I, Brinkman TM, Wang M, et al.: Neurocognitive and psychosocial outcomes in adult survivors of childhood soft-tissue sarcoma: A report from the St. Jude Lifetime Cohort. Cancer 126 (7): 1576-1584, 2020. [PUBMED Abstract]
Tonning Olsson I, Brinkman TM, Hyun G, et al.: Neurocognitive outcomes in long-term survivors of Wilms tumor: a report from the St. Jude Lifetime Cohort. J Cancer Surviv 13 (4): 570-579, 2019. [PUBMED Abstract]
Willard VW, Qaddoumi I, Chen S, et al.: Developmental and adaptive functioning in children with retinoblastoma: a longitudinal investigation. J Clin Oncol 32 (25): 2788-93, 2014. [PUBMED Abstract]
Willard VW, Qaddoumi I, Pan H, et al.: Cognitive and Adaptive Functioning in Youth With Retinoblastoma: A Longitudinal Investigation Through 10 Years of Age. J Clin Oncol 39 (24): 2676-2684, 2021. [PUBMED Abstract]
Brinkman TM, Merchant TE, Li Z, et al.: Cognitive function and social attainment in adult survivors of retinoblastoma: a report from the St. Jude Lifetime Cohort Study. Cancer 121 (1): 123-31, 2015. [PUBMED Abstract]
Ehrhardt MJ, Sandlund JT, Zhang N, et al.: Late outcomes of adult survivors of childhood non-Hodgkin lymphoma: A report from the St. Jude Lifetime Cohort Study. Pediatr Blood Cancer 64 (6): , 2017. [PUBMED Abstract]
Krull KR, Sabin ND, Reddick WE, et al.: Neurocognitive function and CNS integrity in adult survivors of childhood hodgkin lymphoma. J Clin Oncol 30 (29): 3618-24, 2012. [PUBMED Abstract]
Hesko C, Liu W, Srivastava DK, et al.: Neurocognitive outcomes in adult survivors of neuroblastoma: A report from the Childhood Cancer Survivor Study. Cancer 129 (18): 2904-2914, 2023. [PUBMED Abstract]
Wu NL, Phipps AI, Krull KR, et al.: Long-term patient-reported neurocognitive outcomes in adult survivors of hematopoietic cell transplant. Blood Adv 6 (14): 4347-4356, 2022. [PUBMED Abstract]
Willard VW, Leung W, Huang Q, et al.: Cognitive outcome after pediatric stem-cell transplantation: impact of age and total-body irradiation. J Clin Oncol 32 (35): 3982-8, 2014. [PUBMED Abstract]
Kenborg L, Winther JF, Linnet KM, et al.: Neurologic disorders in 4858 survivors of central nervous system tumors in childhood-an Adult Life after Childhood Cancer in Scandinavia (ALiCCS) study. Neuro Oncol 21 (1): 125-136, 2019. [PUBMED Abstract]
Wells EM, Ullrich NJ, Seidel K, et al.: Longitudinal assessment of late-onset neurologic conditions in survivors of childhood central nervous system tumors: a Childhood Cancer Survivor Study report. Neuro Oncol 20 (1): 132-142, 2018. [PUBMED Abstract]
Goldsby RE, Liu Q, Nathan PC, et al.: Late-occurring neurologic sequelae in adult survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. J Clin Oncol 28 (2): 324-31, 2010. [PUBMED Abstract]
Phillips NS, Khan RB, Li C, et al.: Seizures' impact on cognition and quality of life in childhood cancer survivors. Cancer 128 (1): 180-191, 2022. [PUBMED Abstract]
Jain P, Gulati S, Seth R, et al.: Vincristine-induced neuropathy in childhood ALL (acute lymphoblastic leukemia) survivors: prevalence and electrophysiological characteristics. J Child Neurol 29 (7): 932-7, 2014. [PUBMED Abstract]
Ness KK, Jones KE, Smith WA, et al.: Chemotherapy-related neuropathic symptoms and functional impairment in adult survivors of extracranial solid tumors of childhood: results from the St. Jude Lifetime Cohort Study. Arch Phys Med Rehabil 94 (8): 1451-7, 2013. [PUBMED Abstract]
Kandula T, Farrar MA, Cohn RJ, et al.: Chemotherapy-Induced Peripheral Neuropathy in Long-term Survivors of Childhood Cancer: Clinical, Neurophysiological, Functional, and Patient-Reported Outcomes. JAMA Neurol 75 (8): 980-988, 2018. [PUBMED Abstract]
Rodwin RL, Ross WL, Rotatori J, et al.: Newly identified chemotherapy-induced peripheral neuropathy in a childhood cancer survivorship clinic. Pediatr Blood Cancer 69 (3): e29550, 2022. [PUBMED Abstract]
Varedi M, Lu L, Howell CR, et al.: Peripheral Neuropathy, Sensory Processing, and Balance in Survivors of Acute Lymphoblastic Leukemia. J Clin Oncol 36 (22): 2315-2322, 2018. [PUBMED Abstract]
Gurney JG, Kadan-Lottick NS, Packer RJ, et al.: Endocrine and cardiovascular late effects among adult survivors of childhood brain tumors: Childhood Cancer Survivor Study. Cancer 97 (3): 663-73, 2003. [PUBMED Abstract]
Ullrich NJ, Robertson R, Kinnamon DD, et al.: Moyamoya following cranial irradiation for primary brain tumors in children. Neurology 68 (12): 932-8, 2007. [PUBMED Abstract]
Wang C, Roberts KB, Bindra RS, et al.: Delayed cerebral vasculopathy following cranial radiation therapy for pediatric tumors. Pediatr Neurol 50 (6): 549-56, 2014. [PUBMED Abstract]
Mueller S, Fullerton HJ, Stratton K, et al.: Radiation, atherosclerotic risk factors, and stroke risk in survivors of pediatric cancer: a report from the Childhood Cancer Survivor Study. Int J Radiat Oncol Biol Phys 86 (4): 649-55, 2013. [PUBMED Abstract]
Reulen RC, Guha J, Bright CJ, et al.: Risk of cerebrovascular disease among 13 457 five-year survivors of childhood cancer: A population-based cohort study. Int J Cancer 148 (3): 572-583, 2021. [PUBMED Abstract]
Fullerton HJ, Stratton K, Mueller S, et al.: Recurrent stroke in childhood cancer survivors. Neurology 85 (12): 1056-64, 2015. [PUBMED Abstract]
Waxer JF, Wong K, Modiri A, et al.: Risk of Cerebrovascular Events Among Childhood and Adolescent Patients Receiving Cranial Radiation Therapy: A PENTEC Normal Tissue Outcomes Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 417-430, 2024. [PUBMED Abstract]
Khan RB, Merchant TE, Sadighi ZS, et al.: Prevalence, risk factors, and response to treatment for hypersomnia of central origin in survivors of childhood brain tumors. J Neurooncol 136 (2): 379-384, 2018. [PUBMED Abstract]
van Kooten JAMC, Maurice-Stam H, Schouten AYN, et al.: High occurrence of sleep problems in survivors of a childhood brain tumor with neurocognitive complaints: The association with psychosocial and behavioral executive functioning. Pediatr Blood Cancer 66 (11): e27947, 2019. [PUBMED Abstract]
Khan RB, Hudson MM, Ledet DS, et al.: Neurologic morbidity and quality of life in survivors of childhood acute lymphoblastic leukemia: a prospective cross-sectional study. J Cancer Surviv 8 (4): 688-96, 2014. [PUBMED Abstract]
Khong PL, Leung LH, Fung AS, et al.: White matter anisotropy in post-treatment childhood cancer survivors: preliminary evidence of association with neurocognitive function. J Clin Oncol 24 (6): 884-90, 2006. [PUBMED Abstract]
Zeller B, Tamnes CK, Kanellopoulos A, et al.: Reduced neuroanatomic volumes in long-term survivors of childhood acute lymphoblastic leukemia. J Clin Oncol 31 (17): 2078-85, 2013. [PUBMED Abstract]
Faraci M, Morana G, Bagnasco F, et al.: Magnetic resonance imaging in childhood leukemia survivors treated with cranial radiotherapy: a cross sectional, single center study. Pediatr Blood Cancer 57 (2): 240-6, 2011. [PUBMED Abstract]
Phillips NS, Hillenbrand CM, Mitrea BG, et al.: Cerebral microbleeds in adult survivors of childhood acute lymphoblastic leukemia treated with cranial radiation. Sci Rep 10 (1): 692, 2020. [PUBMED Abstract]
Rodwin RL, Chen Y, Yasui Y, et al.: Longitudinal Evaluation of Neuromuscular Dysfunction in Long-term Survivors of Childhood Cancer: A Report from the Childhood Cancer Survivor Study. Cancer Epidemiol Biomarkers Prev 30 (8): 1536-1545, 2021. [PUBMED Abstract]
Rodwin RL, Wang F, Lu L, et al.: Motor and sensory impairment in survivors of childhood central nervous system (CNS) tumors in the St. Jude Lifetime Cohort (SJLIFE). Cancer Med 13 (14): e7422, 2024. [PUBMED Abstract]
Cooper BT, Mayo CS, Milano MT, et al.: Predictive Factors Associated With Radiation Myelopathy in Pediatric Patients With Cancer: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 494-506, 2024. [PUBMED Abstract]
Ness KK, Hudson MM, Jones KE, et al.: Effect of Temporal Changes in Therapeutic Exposure on Self-reported Health Status in Childhood Cancer Survivors. Ann Intern Med 166 (2): 89-98, 2017. [PUBMED Abstract]
Ernst M, Hinz A, Brähler E, et al.: Quality of life after pediatric cancer: comparison of long-term childhood cancer survivors' quality of life with a representative general population sample and associations with physical health and risk indicators. Health Qual Life Outcomes 21 (1): 65, 2023. [PUBMED Abstract]
Schulte F, Brinkman TM, Li C, et al.: Social adjustment in adolescent survivors of pediatric central nervous system tumors: A report from the Childhood Cancer Survivor Study. Cancer 124 (17): 3596-3608, 2018. [PUBMED Abstract]
Vuotto SC, Wang M, Okcu MF, et al.: Neurologic morbidity and functional independence in adult survivors of childhood cancer. Ann Clin Transl Neurol 11 (2): 291-301, 2024. [PUBMED Abstract]
Hörnquist L, Rickardsson J, Lannering B, et al.: Altered self-perception in adult survivors treated for a CNS tumor in childhood or adolescence: population-based outcomes compared with the general population. Neuro Oncol 17 (5): 733-40, 2015. [PUBMED Abstract]
Schulte F, Barrera M: Social competence in childhood brain tumor survivors: a comprehensive review. Support Care Cancer 18 (12): 1499-513, 2010. [PUBMED Abstract]
Papini C, Willard VW, Gajjar A, et al.: Social cognition and adjustment in adult survivors of pediatric central nervous system tumors. Cancer 129 (19): 3064-3075, 2023. [PUBMED Abstract]
Bonneau J, Berbis J, Michel G, et al.: Adolescence and Socioeconomic Factors: Key Factors in the Long-Term Impact of Leukemia on Scholastic Performance-A LEA Study. J Pediatr 205: 168-175.e2, 2019. [PUBMED Abstract]
Drabbe C, Coenraadts ES, van Houdt WJ, et al.: Impaired social functioning in adolescent and young adult sarcoma survivors: Prevalence and risk factors. Cancer 129 (9): 1419-1431, 2023. [PUBMED Abstract]
Ernst M, Brähler E, Wild PS, et al.: Risk factors for suicidal ideation in a large, registry-based sample of adult long-term childhood cancer survivors. J Affect Disord 265: 351-356, 2020. [PUBMED Abstract]
Lubas MM, Mirzaei Salehabadi S, Lavecchia J, et al.: Suicidality among adult survivors of childhood cancer: A report from the St. Jude Lifetime Cohort Study. Cancer 126 (24): 5347-5355, 2020. [PUBMED Abstract]
Brinkman TM, Zhang N, Recklitis CJ, et al.: Suicide ideation and associated mortality in adult survivors of childhood cancer. Cancer 120 (2): 271-7, 2014. [PUBMED Abstract]
Gunnes MW, Lie RT, Bjørge T, et al.: Suicide and violent deaths in survivors of cancer in childhood, adolescence and young adulthood-A national cohort study. Int J Cancer 140 (3): 575-580, 2017. [PUBMED Abstract]
Sun CL, Francisco L, Baker KS, et al.: Adverse psychological outcomes in long-term survivors of hematopoietic cell transplantation: a report from the Bone Marrow Transplant Survivor Study (BMTSS). Blood 118 (17): 4723-31, 2011. [PUBMED Abstract]
Vuotto SC, Krull KR, Li C, et al.: Impact of chronic disease on emotional distress in adult survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 123 (3): 521-528, 2017. [PUBMED Abstract]
Zheng DJ, Krull KR, Chen Y, et al.: Long-term psychological and educational outcomes for survivors of neuroblastoma: A report from the Childhood Cancer Survivor Study. Cancer 124 (15): 3220-3230, 2018. [PUBMED Abstract]
Recklitis C, O'Leary T, Diller L: Utility of routine psychological screening in the childhood cancer survivor clinic. J Clin Oncol 21 (5): 787-92, 2003. [PUBMED Abstract]
Hsu TW, Liang CS, Tsai SJ, et al.: Risk of Major Psychiatric Disorders Among Children and Adolescents Surviving Malignancies: A Nationwide Longitudinal Study. J Clin Oncol 41 (11): 2054-2066, 2023. [PUBMED Abstract]
De R, Sutradhar R, Kurdyak P, et al.: Incidence and Predictors of Mental Health Outcomes Among Survivors of Adolescent and Young Adult Cancer: A Population-Based Study Using the IMPACT Cohort. J Clin Oncol 39 (9): 1010-1019, 2021. [PUBMED Abstract]
Phipps S, Klosky JL, Long A, et al.: Posttraumatic stress and psychological growth in children with cancer: has the traumatic impact of cancer been overestimated? J Clin Oncol 32 (7): 641-6, 2014. [PUBMED Abstract]
Stuber ML, Meeske KA, Leisenring W, et al.: Defining medical posttraumatic stress among young adult survivors in the Childhood Cancer Survivor Study. Gen Hosp Psychiatry 33 (4): 347-53, 2011 Jul-Aug. [PUBMED Abstract]
Phipps S, Larson S, Long A, et al.: Adaptive style and symptoms of posttraumatic stress in children with cancer and their parents. J Pediatr Psychol 31 (3): 298-309, 2006. [PUBMED Abstract]
Phipps S, Jurbergs N, Long A: Symptoms of post-traumatic stress in children with cancer: does personality trump health status? Psychooncology 18 (9): 992-1002, 2009. [PUBMED Abstract]
Rourke MT, Hobbie WL, Schwartz L, et al.: Posttraumatic stress disorder (PTSD) in young adult survivors of childhood cancer. Pediatr Blood Cancer 49 (2): 177-82, 2007. [PUBMED Abstract]
Schwartz L, Drotar D: Posttraumatic stress and related impairment in survivors of childhood cancer in early adulthood compared to healthy peers. J Pediatr Psychol 31 (4): 356-66, 2006. [PUBMED Abstract]
Stuber ML, Meeske KA, Krull KR, et al.: Prevalence and predictors of posttraumatic stress disorder in adult survivors of childhood cancer. Pediatrics 125 (5): e1124-34, 2010. [PUBMED Abstract]
Hobbie WL, Stuber M, Meeske K, et al.: Symptoms of posttraumatic stress in young adult survivors of childhood cancer. J Clin Oncol 18 (24): 4060-6, 2000. [PUBMED Abstract]
Dieluweit U, Debatin KM, Grabow D, et al.: Social outcomes of long-term survivors of adolescent cancer. Psychooncology 19 (12): 1277-84, 2010. [PUBMED Abstract]
Seitz DC, Hagmann D, Besier T, et al.: Life satisfaction in adult survivors of cancer during adolescence: what contributes to the latter satisfaction with life? Qual Life Res 20 (2): 225-36, 2011. [PUBMED Abstract]
Tai E, Buchanan N, Townsend J, et al.: Health status of adolescent and young adult cancer survivors. Cancer 118 (19): 4884-91, 2012. [PUBMED Abstract]
Schultz KA, Ness KK, Whitton J, et al.: Behavioral and social outcomes in adolescent survivors of childhood cancer: a report from the childhood cancer survivor study. J Clin Oncol 25 (24): 3649-56, 2007. [PUBMED Abstract]
Prasad PK, Hardy KK, Zhang N, et al.: Psychosocial and Neurocognitive Outcomes in Adult Survivors of Adolescent and Early Young Adult Cancer: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 33 (23): 2545-52, 2015. [PUBMED Abstract]
Brinkman TM, Li C, Vannatta K, et al.: Behavioral, Social, and Emotional Symptom Comorbidities and Profiles in Adolescent Survivors of Childhood Cancer: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 34 (28): 3417-25, 2016. [PUBMED Abstract]
Karlson CW, Alberts NM, Liu W, et al.: Longitudinal pain and pain interference in long-term survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 126 (12): 2915-2923, 2020. [PUBMED Abstract]
Tonning Olsson I, Alberts NM, Li C, et al.: Pain and functional outcomes in adult survivors of childhood cancer: A report from the St. Jude Lifetime Cohort study. Cancer 127 (10): 1679-1689, 2021. [PUBMED Abstract]
Frederiksen LE, Erdmann F, Mader L, et al.: Psychiatric disorders in childhood cancer survivors in Denmark, Finland, and Sweden: a register-based cohort study from the SALiCCS research programme. Lancet Psychiatry 9 (1): 35-45, 2022. [PUBMED Abstract]
Papini C, Mirzaei S S, Xing M, et al.: Evolving therapies, neurocognitive outcomes, and functional independence in adult survivors of childhood glioma. J Natl Cancer Inst 116 (2): 288-298, 2024. [PUBMED Abstract]
Efectos tardíos en el aparato digestivo
Dentales
Aspectos generales
Es posible que la quimioterapia, la radioterapia y la cirugía produzcan varias anomalías cosméticas y funcionales en la cavidad oral y la dentadura. La calidad de la evidencia científica vigente relacionada con este desenlace es limitada debido a la recopilación retrospectiva de datos, las muestras pequeñas, el sesgo de selección de la cohorte y el sesgo de participación, y la heterogeneidad en el abordaje de tratamiento, el tiempo trascurrido desde el tratamiento y el método de comprobación.
Las complicaciones orales y dentales que se notifican en los sobrevivientes de cáncer infantil son las siguientes:
Las anomalías notificadas en el desarrollo de la dentición en sobrevivientes de cáncer infantil son las siguientes:[1-12]
Falta de desarrollo de la dentición.
Microdoncia.
Hipoplasia adamantina.
Deformidad radicular.
Hipodoncia.
La prevalencia de hipodoncia ha variado mucho en las series, según la edad en el momento del diagnóstico, la modalidad de tratamiento y el método de comprobación.
Los tratamientos para el cáncer que se relacionaron con defectos en el desarrollo dental son los siguientes:[3,10]
Los niños menores de 5 años tienen un mayor riesgo de presentar anomalías de la dentición, como agenesia radicular, erupción demorada, defectos adamantinos o exceso de caries relacionadas con una alteración de la actividad de los ameloblastos (productores de esmalte) y los odontoblastos (productores de dentina) que se produce al principio de la vida.[3]
Los siguientes son los hallazgos clave relacionados con el efecto del tratamiento del cáncer en el desarrollo de la dentición:
Radioterapia
La radiación dirigida a la cavidad oral o las estructuras que la rodean aumenta el riesgo de anomalías de la dentición porque los ameloblastos se pueden dañar de modo permanente con dosis de tan solo 10 Gy. Además, la exposición de las glándulas mayores y menores repercute en la función salival.[3-5]
El grado más importante de aplasia dental o erupción demorada se presenta en los niños más pequeños (<4 años) expuestos a dosis de radiación de 20 Gy o más.[13]
En ocasiones, los dientes en desarrollo se irradian durante el tratamiento de sarcomas de cabeza y cuello, linfoma de Hodgkin, neuroblastoma, leucemia en el sistema nervioso central, cáncer de nasofaringe y tumores de encéfalo, y durante la irradiación corporal total (ICT) para un TCMH.
Las dosis de 10 a 40 Gy a veces causan acortamiento de las raíces o curvatura anormal, enanismo e hipocalcificación.[14]
Se notificaron anomalías de la dentición significativas, como hipoplasia mandibular o maxilar, aumento del número de caries, hipodoncia, microdoncia, atrofia de las raíces y xerostomía en más del 85 % de los sobrevivientes de rabdomiosarcomas de cabeza y cuello tratados con dosis de radiación superiores a 40 Gy.[4,15]
En la cohorte St. Jude Life (SJLIFE) los sobrevivientes que recibieron radioterapia, que potencialmente expuso la cavidad oral, tuvieron más probabilidad de informar al menos un problema de salud dental (oportunidad relativa [OR], 1,48; intervalo de confianza [IC] 95 %, 1,26–1,72) y al menos una anomalía de tejido blando (OR, 1,52; IC 95 %, 1,25–1,84) después del control por factores socioeconómicos, edad en el último seguimiento y diagnóstico, otras exposiciones al tratamiento y acceso a servicios dentales.[12]
Quimioterapia
La quimioterapia, en particular la exposición a alquilantes, puede afectar el desarrollo de la dentición.[3,5,6]
La quimioterapia para el tratamiento de la leucemia o el neuroblastoma se vincula con acortamiento y adelgazamiento de las raíces premolares y anormalidades en el esmalte.[8,16,17]
En el Childhood Cancer Survivor Study (CCSS), los investigadores identificaron la edad menor de 5 años y un aumento de la exposición a la ciclofosfamida como factores de riesgo importante de anomalías en el desarrollo de la dentición en sobrevivientes a largo plazo de cáncer infantil.[3]
En el estudio SJLIFE, los sobrevivientes que recibieron alquilantes clásicos (OR, 1,6; IC 95 %, 1,36–1,88) o antibióticos tipo antraciclinas (OR, 1,22; IC 95 %, 1,04–1,42) tenían más probabilidad de notificar al menos un problema de salud dental después de controlar por factores socioeconómicos, edad en el último seguimiento y diagnóstico, otras exposiciones al tratamiento y acceso a servicios dentales.[12]
Trasplante de células madre hematopoyéticas
Es posible que el acondicionamiento para un trasplante de células madre hematopoyéticas (TCMH), en particular los regímenes con ICT, produzcan agenesia dental y malformaciones en las raíces.[1,2]
Los niños más pequeños que no han desarrollado la dentición secundaria son los más vulnerables.[1,2,5]
Los niños que se someten a TCMH con ICT a veces presentan raíces en forma de V, microdoncia, hipoplasia adamantina o cierre apical prematuro.[1,2,7]
Una edad más joven en el momento del TCMH se vincula con problemas más graves de malformación dental y menor crecimiento vertical de la parte inferior de la cara.[8]
Las anomalías de la dentición se notificaron en pacientes que recibieron TCMH sin ICT, en particular en pacientes menores de 2 años en el momento del trasplante.[17]
Disfunción de las glándulas salivales
La xerostomía, sensación de sequedad en la boca, es un efecto secundario posible después de la irradiación de cabeza y cuello o el TCMH que a veces afecta de forma grave la calidad de vida.[18]
Las complicaciones producidas por la reducción de la secreción de saliva son las siguientes:[18,19]
Aumento de caries.
Susceptibilidad a infecciones orales.
Alteraciones del sueño.
Dificultades para masticar, tragar y hablar.
La prevalencia de la disfunción de las glándulas salivales después del tratamiento del cáncer varía de acuerdo con las técnicas de medición (notificación del paciente vs. tasas de secreción salival estimulada o no estimulada).[20]
En general, la prevalencia de xerostomía persistente autonotificada después del tratamiento suele ser baja en los sobrevivientes de cáncer infantil.
En el CCSS, la prevalencia de xerostomía autonotificada por los sobrevivientes fue del 2,8 %, en comparación con el 0,3 % en los hermanos, con un aumento de riesgo en los sobrevivientes mayores de 30 años.[3]
Los hallazgos clave relacionados con el efecto del tratamiento del cáncer en el funcionamiento de las glándulas salivales son los siguientes:
Radioterapia
La irradiación accidental de las glándulas salivales relacionada con el tratamiento de neoplasias malignas de cabeza y cuello o linfoma de Hodgkin genera un cambio cualitativo y cuantitativo en el flujo salival, que a veces se revierte después de dosis inferiores a 40 Gy, pero que tal vez sea irreversible después de dosis más altas, según se administre también quimioterapia sensibilizante.[18]
En un estudio alemán de 114 pacientes pediátricos, el riesgo de xerostomía aguda y tardía aumentó según la dosis dirigida a la glándula parótida y submandibular. En general, se observó xerostomía de grado 1 o superior en pacientes que recibieron una dosis máxima superior a 20 Gy en las glándulas salivales. Las probabilidades de presentar xerostomía aguda y tardía fueron más altas en los pacientes que recibieron quimioterapia simultánea en comparación con los que recibieron radioterapia sola, con una OR de 3,64 (IC 95 %, 1,49–8,89) para la xerostomía aguda y de 5,15 (IC 95 %, 1,20–22,15) para la xerostomía tardía.[21]
La iniciativa Pediatric Normal Tissue Effects in the Clinic (PENTEC) notificó los efectos tardíos en los sobrevivientes de cáncer infantil tratados con radioterapia para los cánceres de cabeza y cuello.[22] Se observó xerostomía de grado 1 o superior en pacientes que recibieron una dosis máxima de más de 20 Gy en las glándulas salivales. La probabilidad de xerostomía aguda y crónica fue mayor en los pacientes que habían recibido quimioterapia y radioterapia simultáneas (OR de xerostomía aguda, 3,64; IC 95 %, 1,49–8,89 y xerostomía tardía OR, 5,15; IC 95 %, 1,20–22,15), en comparación con los pacientes tratados con radioterapia sola. Una dosis media parotídea de 35 a 40 Gy se relacionó con un riesgo del 32 % de presentar xerostomía aguda superior a grado 2 y un riesgo del 13 % al 32 % de xerostomía crónica.
Trasplante de células madre hematopoyéticas
Los receptores de trasplante de células madre hematopoyéticas (TCMH) tienen un aumento de riesgo de disfunción de las glándulas salivales relacionada con el acondicionamiento para el trasplante o la enfermedad de injerto contra huésped (EICH).
La EICH puede producir hiposalivación y xerostomía con la enfermedad dental resultante.
En un estudio de sobrevivientes de EICH infantil, el 60 % de aquellos expuestos a un régimen de acondicionamiento con ciclofosfamida y 10 Gy de ICT en una sola dosis tuvieron una disminución de las tasas de secreción salival, en comparación con el 26 % de aquellos que recibieron ciclofosfamida y busulfano.[23]
En contraste, en otro estudio, la prevalencia de reducción de secreción salival no difirió entre los sobrevivientes a largo plazo según el régimen de acondicionamiento (ICT en dosis única, 47 %; ICT fraccionada, 47 %; busulfano, 42 %).[24]
Quimioterapia
La relación de la quimioterapia sola con la xerostomía continúa siendo objeto de polémica.[18]
Solo en un estudio de pacientes pediátricos se demostró un exceso de riesgo (OR, 12,32, IC 95 %, 2,1–74,4) de disminución del flujo de saliva estimulado en pacientes tratados con ciclofosfamida. Sin embargo, no se observó un aumento de caries dental y no se evaluó la xerostomía notificada por el paciente.[6]
Anomalías en el desarrollo craneofacial
La alteración del desarrollo craneofacial es una consecuencia adversa frecuente en los niños tratados con dosis altas de radioterapia dirigida a la cabeza y el cuello, que se suele presentar en relación con otras secuelas en la cavidad oral, como anomalías dentales, xerostomía y trismo.[4,25]
El grado y la gravedad de desfiguramiento osteomuscular se relaciona con la edad en el momento del tratamiento, así como el volumen y la dosis de la radioterapia; el riesgo es más alto en los pacientes más jóvenes y en aquellos que recibieron radioterapia de 30 Gy o más.[25]
Otras complicaciones orales
La osteorradionecrosis en la mandíbula es una complicación infrecuente en los sobrevivientes de cáncer infantil tratados con dosis altas de radiación craneofacial (>40 Gy), en particular después de extracciones dentales en mandíbulas irradiadas.[26,27]
Suelen ser necesarias varias intervenciones quirúrgicas para corregir las anomalías cosméticas y funcionales.
Se desconoce el efecto de las complicaciones y las alteraciones infecciosas en la microflora durante el tratamiento y después de este.[5]
Atención posterior al tratamiento
En algunos estudios, se indica que los productos con fluoruro o los enjuagues con clorhexidina tal vez sean beneficiosos para los pacientes que se sometieron a radioterapia.[28]
La caries dental es una consecuencia problemática de la reducción de la calidad y el flujo de saliva. El uso de fluoruro tópico reduce de manera considerable la frecuencia de las caries, y los sustitutos de saliva y los sialagogos mejoran secuelas como la xerostomía.[19]
En el Cuadro 5 se resumen los efectos tardíos orales y dentales, y los exámenes médicos de detección correspondientes.
Cuadro 5. Efectos tardíos orales o dentalesa
Tratamiento predisponente
Efectos orales o dentales
Exámenes médicos de detección e intervenciones
TC = tomografía computarizada; EICH = enfermedad de injerto contra huésped; TCMH = trasplante de células madre hematopoyéticas; IRM = imágenes por resonancia magnética.
Cualquier tipo de quimioterapia; radioterapia con exposición de la cavidad oral
Anomalías del desarrollo dental; agenesia de dientes o raíces; microdoncia; adelgazamiento o acortamiento de las raíces; displasia adamantina
Evaluación y limpieza dentales cada 6 meses
Atención dental habitual que incluya aplicaciones de fluoruro
Consulta con un ortodoncista especializado en el tratamiento de sobrevivientes de cáncer infantil irradiados
Radiografía panorámica (Panorex) inicial antes de los procedimientos dentales para evaluar el desarrollo de las raíces
Radioterapia con exposición de la cavidad oral
Oclusión dental defectuosa y disfunción de la articulación temporomandibular
Evaluación y limpieza dentales cada 6 meses
Atención dental habitual que incluya aplicaciones de fluoruro
Consulta con un ortodoncista especializado en el tratamiento de sobrevivientes de cáncer infantil irradiados
Radiografía panorámica (Panorex) inicial antes de los procedimientos dentales para evaluar el desarrollo de las raíces
Derivación a un otorrinolaringólogo para obtener dispositivos auxiliares para la apertura de la mandíbula
Radioterapia con exposición de la cavidad oral; TCHM con antecedentes de EICH crónica
Xerostomía o disfunción de las glándulas salivales; enfermedad periodontal; caries; cáncer oral (carcinoma de células escamosas)
Evaluación y limpieza dentales cada 6 meses
Tratamiento complementario con sustitutos de saliva, hidratantes y sialagogos (pilocarpina)
Atención dental habitual que incluya aplicaciones de fluoruro
Derivación para biopsia de lesiones sospechosas.
Radioterapia con exposición de la cavidad oral (≥40 Gy)
Osteorradionecrosis
Antecedentes: curación deficiente o tardía después de procedimientos dentales
Hallazgos del examen: dolor maxilar persistente, hinchazón o trismo
Los estudios con imágenes (radiografía, TC o IRM) quizás sean útiles para el diagnóstico
A veces se necesita una biopsia quirúrgica para confirmar el diagnóstico
Considerar el tratamiento con oxígeno hiperbárico
Tubo digestivo
Aspectos generales
El tubo digestivo es sensible a los efectos tóxicos agudos de la quimioterapia, la radioterapia y la cirugía. Estas modalidades importantes de tratamiento también causan problemas a largo plazo según el tratamiento y la dosis.
Los informes publicados sobre los desenlaces a largo plazo en el tubo digestivo se ven limitados debido a la recopilación retrospectiva de datos, el tamaño pequeño de la muestra, el sesgo de selección y participación de la cohorte, la heterogeneidad en el abordaje de tratamiento, el tiempo transcurrido desde el tratamiento y el método de confirmación.
Los efectos tardíos relacionados con el tratamiento son los siguientes:
Efectos tardíos en el tubo digestivo superior e inferior relacionados con la intensidad de la dosis de quimioterapia o de radiación abdominal.
Adherencias posteriores a la cirugía abdominal que predisponen a la obstrucción intestinal posoperatoria.
Los siguientes son los efectos tardíos relacionados con el tubo digestivo:
Dismotilidad esofágica.
Estenosis esofágica.
Reflujo gastroesofágico.
Gastritis, enteritis o colitis.
Disfunción de la motilidad del tubo digestivo (diarrea, estreñimiento, encopresis, obstrucción intestinal).
Neoplasias malignas subsiguientes (NMS).
Efectos del tipo histológico del cáncer en los desenlaces en el tubo digestivo
El abdomen representa una localización relativamente común de varias neoplasias malignas infantiles, como el rabdomiosarcoma, el tumor de Wilms, el linfoma, los tumores de células germinativas y el neuroblastoma.
Los tumores intraabdominales a menudo necesitan tratamiento multimodal, a veces resección del intestino, quimioterapia que lesiona el intestino o radioterapia. Por lo tanto, se espera que estos tumores sean particularmente propensos a producir problemas a largo plazo en el tubo digestivo.
Desenlaces en el tubo digestivo a partir de estudios de cohortes específicos
Evidencia (desenlaces en el tubo digestivo a partir de estudios de cohortes específicos):
En los sobrevivientes a 5 años de cáncer infantil que participaron en el CCSS, la incidencia acumulada de afecciones digestivas autonotificadas fue del 37,6 % 20 años después del diagnóstico del cáncer (25,8 % por complicaciones digestivas altas y 15,5 % por complicaciones digestivas bajas); estas cifras representan un exceso de riesgo de casi el doble de complicaciones digestivas altas (riesgo relativo [RR], 1,8; IC 95 %, 1,6–2,0) y de complicaciones digestivas bajas (RR, 1,9; IC 95 %, 1,7–2,2), en comparación con los hermanos de control.[29]
Los factores pronósticos de un riesgo más elevado de complicaciones digestivas específicas son los siguientes:
Mayor edad en el momento del diagnóstico.
Intensificación del tratamiento (antraciclinas para complicaciones digestivas altas y alquilantes para complicaciones digestivas bajas).
Radioterapia abdominal.
Cirugía abdominal.
Daños relacionados con la radiación dirigida al tubo digestivo
Los daños tardíos por la radiación dirigida al tubo digestivo se atribuyen a lesión vascular.
Se puede presentar necrosis, úlcera, estenosis o perforación, caracterizadas por hipoabsorción, dolor y episodios repetidos de obstrucción intestinal, así como perforación e infección.[30-32]
En general, es posible administrar las dosis de radiación fraccionada de 20 a 30 Gy al intestino delgado sin morbilidad significativa en el largo plazo.[33]
Las dosis superiores a 40 GY se relacionan con un riesgo más alto de obstrucción intestinal o enterocolitis crónica.[33]
Los fármacos quimioterapéuticos sensibilizantes, como la dactinomicina o las antraciclinas, tal vez aumenten este riesgo.
En un número limitado de informes, se describen complicaciones digestivas en pacientes pediátricos con tumores genitourinarios sólidos tratados con radioterapia:
En el CCSS se evaluó la morbilidad a largo plazo entre los sobrevivientes de tumor de Wilms no sindrómico unilateral (n = 2008) de acuerdo con los regímenes de tratamiento convencionales. Todos los pacientes se sometieron a nefrectomía unilateral.[34]
La incidencia acumulada a 35 años de obstrucción intestinal fue del 8,1 % (IC 95 %, 6,6–9,6 %).
En relación con los hermanos, los sobrevivientes tratados con vincristina y actinomicina D (VA) mostraron un riesgo elevado de obstrucción intestinal (RR, 9,4). El riesgo con el tratamiento de VA fue más bajo que para los grupos de tratamiento que recibieron terapia más intensiva (intervalo RR, 15,4–46,8).
La tasa de obstrucción intestinal fue más alta en los sobrevivientes que recibieron más de 20 Gy de radioterapia completa dirigida al abdomen o al flanco.
En el CCSS se evaluó la incidencia y el riesgo de obstrucción intestinal tardía que exigió tratamiento quirúrgico en 12 316 sobrevivientes a 5 años (2002 con tumores abdominopélvicos y 10 314 sin este tipo de tumores) y 4023 hermanos.[35]
Los diagnósticos más comunes en los sobrevivientes con tumores abdominopélvicos fueron tumor de Wilms y neuroblastomas, pero también se incluyeron sarcomas de tejidos blandos, linfomas y tumores de hueso.
La incidencia acumulada de una obstrucción intestinal tardía que exigió tratamiento quirúrgico a los 35 años fue del 5,8 % en los sobrevivientes de tumores abdominopélvicos, del 1,0 % entre aquellos sin tumores abdominopélvicos y del 0,3 % en los hermanos.
El riesgo alto de obstrucción intestinal que exigió tratamiento quirúrgico se relacionó con la presencia de un tumor abdominopélvico (razón de tasas ajustada [RTA], 3,6; P < 0,001) y la exposición a la radioterapia dirigida al abdomen o la pelvis dentro de los 5 años siguientes al diagnóstico de cáncer (RTA, 2,4; P < 0,001).
En los sobrevivientes de tumores abdominopélvicos, la mediana de tiempo desde el diagnóstico hasta la primera obstrucción intestinal tardía que exigió tratamiento quirúrgico fue de 12 años (intervalo, 8–19 años).
El linfoma se relacionó con la incidencia acumulada más alta de obstrucción intestinal tardía que exigió tratamiento quirúrgico (7,2 % a los 35 años del diagnóstico).
En el CCSS se evaluó la enfermedad anorrectal de inicio tardío en miembros de la cohorte:[36]
Entre los sobrevivientes, la radioterapia pélvica superior a 30 Gy dentro de los 5 años del diagnóstico de cáncer se relacionó con enfermedad anorrectal de inicio tardío (CTA para 30–49,9 Gy vs. ninguna, 1,6; CTA para ≥50 Gy vs. ninguna, 5,4).
La enfermedad anorrectal que se notificó con mayor frecuencia fue la fístula, seguida de estenosis y NMS anorrectal.
La enfermedad anorrectal de inicio tardío se relacionó con afectación psicológica en todos los ámbitos, que se caracteriza por un aumento de sufrimiento emocional y deterioro de la calidad de vida.
Según informes del Intergroup Rhabdomyosarcoma Study, en el que se evaluó la toxicidad en el tubo digestivo en sobrevivientes a largo plazo de rabdomiosarcomas genitourinarios, las anomalías en el intestino irradiado fueron infrecuentes.[37-39]
Se presentaron complicaciones relacionadas con la radiación en cerca del 10 % de los sobrevivientes a largo plazo de rabdomiosarcoma paratesticular, y de vejiga o próstata, como adherencias intraperitoneales con obstrucción intestinal, diarrea crónica y formación de estenosis o fístulas intestinales.
El CCSS evaluó la prevalencia de estenosis esofágica e identificó factores de riesgo clínicos y de tratamiento relacionados entre 17 121 sobrevivientes a 5 años de cáncer infantil y 3400 hermanos.[40]
La prevalencia de estenosis esofágica entre los sobrevivientes fue del 2,0 % (IC 95 %, 1,8–2,2 %), lo que representa un riesgo 7,6 veces mayor, en comparación con los hermanos.
Los factores que se relacionaron de manera significativa con el riesgo de estenosis esofágica fueron el diagnóstico de linfoma de Hodgkin, dosis más altas de radiación torácica, edad más temprana en el momento del diagnóstico del cáncer, exposición a la quimioterapia con derivados del platino y someterse a un TCMH.
En el Cuadro 6 se resumen los efectos tardíos en el tubo digestivo y los exámenes médicos de detección correspondientes.
Cuadro 6. Efectos tardíos en el tubo digestivoa
Tratamiento predisponente
Efectos digestivos
Exámenes médicos de detección e intervenciones
EICH = enfermedad de injerto contra huésped; TCMH = trasplante de células madre hematopoyéticas; RUV= riñones, uréteres, vejiga (radiografía simple del abdomen).
Dilatación del esófago, tratamiento médico y cirugía antirreflujo
Radioterapia con exposición del intestino
Enterocolitis crónica, fístulas, estenosis
Antecedentes: náuseas, vómitos, dolor abdominal y diarrea
Concentraciones séricas de proteínas y albúmina una vez al año para pacientes con diarrea crónica o fístulas; consulta gastroenterológica
Consulta quirúrgica o gastroenterológica para pacientes sintomáticos
Radioterapia con exposición del intestino; laparotomía
Obstrucción intestinal
Antecedentes: dolor abdominal, distensión, vómitos y estreñimiento
Hallazgos del examen: sensibilidad a la palpación, defensa abdominal y distensión (episodio agudo)
Evaluación clínica en pacientes con síntomas clínicos de obstrucción
Consulta quirúrgica para pacientes que no responden al tratamiento clínico
Cirugía pélvica y cistectomía
Incontinencia fecal
Antecedentes: estreñimiento crónico e incontinencia fecal
Examen del recto
Hepatobiliares
Aspectos generales
Las complicaciones hepáticas del tratamiento del cáncer infantil se observan principalmente como toxicidad aguda en respuesta al tratamiento.[41] Debido a que muchos fármacos quimioterapéuticos y la radiación son hepatotóxicos, las anomalías transitorias de la función hepática son comunes durante el tratamiento. Las complicaciones hepáticas graves son infrecuentes. Los sobrevivientes de cáncer infantil presentan, en ocasiones, una lesión hepática prolongada.[42]
Algunos conceptos generales acerca de la hepatotoxicidad relacionada con el cáncer infantil son los siguientes:
No está bien definido el riesgo de hepatotoxicidad a largo plazo.
Los niños con tumores hepáticos primarios que exigen una resección hepática importante o incluso un trasplante, tienen riesgo más alto de lesiones hepáticas.
Los niños sometidos a radioterapia dirigida al hígado tienen riesgo más alto de lesiones hepáticas.
Los niños que se someten a un trasplante de médula ósea tienen un riesgo más alto de lesiones hepáticas.
Ciertos factores, como el tipo de quimioterapia, la dosis y el grado de exposición a la radiación, la influencia de intervenciones quirúrgicas y el efecto progresivo de una hepatitis viral u otras complicaciones infecciosas, exigen atención adicional en futuros estudios.
Tipos de efectos tardíos hepatobiliares
Las elevación asintomática de enzimas hepáticas es la complicación hepatobiliar más común.
Elevación asintomática de enzimas hepáticas. La lesión hepática relacionada con el tratamiento del cáncer infantil es, a menudo, asintomática y de progreso lento. Si bien las concentraciones elevadas de alanina–aminotransferasa (ALT), aspartato–aminotransferasa (AST) y γ-glutamil–transferasa (γ-GT) pueden reflejar una lesión hepática aguda transitoria durante la quimioterapia, no pronostican disfunción hepática tardía o cirrosis.
Investigadores neerlandeses observaron disfunción hepatobiliar en el 8,7 % de 1362 sobrevivientes a largo plazo (mediana de seguimiento 12,4 años desde el diagnóstico) evaluada mediante ALT para la lesión hepatocelular y mediante GGT para lesiones de las vías biliares. Se excluyeron los casos con antecedentes de hepatitis vírica y enfermedad venoclusiva.[43]
Los factores de predicción de concentraciones elevadas de ALT y γ-GT evaluados por medio de análisis multivariantes incluyeron la radioterapia con exposición del hígado, un índice de masa corporal (IMC) más alto, un mayor consumo de bebidas alcohólicas y el tiempo de seguimiento más prolongado; la edad más avanzada en el momento del diagnóstico solo se relacionó de forma significativa con concentraciones elevadas de γ-GT.
En un análisis del estudio SJLIFE se evaluaron la prevalencia y los factores de riesgo de concentraciones elevadas de ALT entre 2751 adultos sobrevivientes de cáncer infantil (mediana de edad, 31,4 años; mediana de tiempo desde el diagnóstico, 23,2 años).[44]
En el 41,3 % de los sobrevivientes predominó una concentración de ALT mayor que los límites superiores de normalidad específicos para el sexo; sin embargo, la prevalencia de daño hepático de grado 3 o 4 fue infrecuente (<1 %).
Los factores de riesgo independiente para la elevación de la ALT incluyeron la raza o la etnia blanca no hispana, la edad más avanzada en el momento de la evaluación, el sobrepeso o la obesidad, la presencia de síndrome metabólico, el tratamiento actual con una estatina, la infección por el virus de la hepatitis C, el tratamiento previo con busulfano o tioguanina, el antecedente de cirugía hepática y el porcentaje del hígado que se trató con dosis de 10 Gy, 15 Gy, 20 Gy, o mayores.
Las complicaciones hepatobiliares que se notificaron menos fueron las siguientes:[45]
Colelitiasis
En pocos estudios, un aumento de riesgo de colelitiasis se relacionó con el conducto ileal, la nutrición parenteral, la cirugía abdominal, la radioterapia abdominal y el TCMH.[46,47]
La incidencia acumulada de colecistectomía tardía (5 o más años después del diagnóstico de cáncer) en 25 549 participantes del CCSS diagnosticados entre 1970 y 1999 (mediana de seguimiento, 21,9 años) fue del 7,2 % en comparación con el 6,6 % en un grupo de control de hermanos (razón de tasas, 1,3; IC 95 %, 1,1–1,5).[48]
Los factores de riesgo independiente para la colecistectomía incluyen la edad, el sexo femenino, un mayor IMC y exposición a dosis altas (>750 mg/m2) de quimioterapia con derivados de platino (razón de tasas, 2,6; IC 95 %, 1,5–4,5), quimioterapia con alcaloide de vinca (razón de tasas, 1,4; IC 95 %, 1,1–1,8) o ICT (cociente de tasas, 2,2; IC 95 %, 1,2–2,4).[48]
Hiperplasia nodular focal
En estudios con imágenes de detección después de la quimioterapia o un TCMH, se descubrieron de forma casual lesiones formadas por la regeneración del hígado, llamadas lesiones por hiperplasia focal nodular (HFN).[49,50]
Se piensa que las HFN son una manifestación iatrogénica benigna de daño vascular, y se relacionaron con enfermedad venoclusiva, dosis altas de alquilantes (por ejemplo, busulfano y melfalán) e irradiación hepática.[49]
Se desconoce la prevalencia de la HFN que, si bien se menciona como inferior al 1 % en algunos artículos,[50] es probable que esté subestimada.
En un estudio de pacientes a quienes se les realizó un seguimiento mediante imágenes por resonancia magnética (IRM) después de un trasplante para evaluar depósitos de hierro en el hígado, la incidencia acumulada de HFN fue del 35 % a los 150 meses del trasplante.[49]
La HFN puede simular tumores metastásicos o subsiguientes, pero en las imágenes con IRM exhiben un modelo característico que suele ser diagnóstico.[49]
Por lo general, no son necesarias la biopsia o la resección a menos que las lesiones crezcan o los pacientes tengan síntomas preocupantes.[49]
Hiperplasia nodular regenerativa
La hiperplasia nodular regenerativa es una afección poco frecuente que se caracteriza por múltiples nódulos hepáticos monoacinares regenerativos y fibrosis leve.[45]
La etiopatogenia de la hiperplasia nodular regenerativa no está bien establecida, pero es posible que represente una adaptación tisular inespecífica a un flujo sanguíneo hepático heterogéneo.[51]
Esta afección se ha observado con poca frecuencia en los sobrevivientes de cáncer infantil tratados con quimioterapia, con irradiación dirigida al hígado o sin esta.[52,53]
A veces se necesita realizar una biopsia para diferenciar una hiperplasia nodular regenerativa de una neoplasia maligna subsiguiente.[53]
Cambio en la grasa microvesicular
Se observaron indicios histológicos de infiltración grasa (93 %) y siderosis (hasta 70 %) en niños con leucemia linfoblástica aguda (LLA) que recién habían completado el tratamiento intensificado. Entre los pacientes, el 11 % presentó fibrosis que se relacionó con una concentración sérica más alta de colesterol de lipoproteínas de baja densidad (LDL).[54]
También se notificó que el hígado graso con resistencia a la insulina se presenta con más frecuencia en sobrevivientes de cáncer infantil a largo plazo tratados con radioterapia craneal e ICT antes de un TCMH alogénico que no tenían sobrepeso ni eran obesos.[55]
Se necesitan estudios prospectivos para definir si el cambio agudo del hígado graso después del tratamiento contribuye a la presentación de esteatohepatitis tardía o síndrome metabólico en esta población.
Hemocromatosis adquirida
La transfusión de glóbulos rojos a veces produce una acumulación del hierro excedente causada por la alteración de la homeostasis del almacenamiento y la distribución de este mineral cuando se acumula hierro exógeno en los órganos.
Se notificó sobrecarga de hierro transfusional en el ámbito de la oncología pediátrica, pero su prevalencia, la distribución en los órganos y la gravedad no se conocen bien.
La IRM se ha convertido en un medio exacto y no invasivo para medir el hierro en múltiples órganos.[56,57]
En un estudio transversal de 103 pacientes (incluso 73 que ya no recibían tratamiento), el 53 % (54 de 102) presentaron siderosis hepática moderada o mayor, el 80 % (77 de 96) siderosis pancreática, el 4 % (3 de 80) siderosis cardiaca y el 45 % (13 de 29) siderosis hipofisaria o pérdida de volumen. Las concentraciones de ferritina fueron más bajas en los pacientes que ya no recibían tratamiento que en los que sí lo recibían (P = 0,0008). La ferritina se relacionó con la concentración hepática de hierro solo en los pacientes que ya no recibían tratamiento (P = 0,0016). La concentración de hierro hepático y pancreático no difirió entre los dos grupos de pacientes. El T2 cardíaco fue ligeramente inferior en los pacientes que seguían recibiendo terapia, pero no a un nivel relevante desde el punto de vista clínico.[58]
La recepción de un trasplante alogénico es un factor de riesgo significativo de sobrecarga de hierro transfusional.[59]
Factores de riesgo relacionados con el tratamiento de los efectos tardíos hepatobiliares
El tipo y la intensidad del tratamiento previo influyen en el riesgo de efectos hepatobiliares tardíos. Además del riesgo de toxicidad relacionada con el tratamiento, los receptores de TCMH presentan con frecuencia disfunción hepática crónica relacionada con causas microvasculares, inmunitarias, infecciosas, metabólicas y otras causas tóxicas.
Los hallazgos clave relacionados con el efecto del tratamiento del cáncer en la presentación de complicaciones hepatobiliares son los siguientes:
Quimioterapia
Los fármacos quimioterapéuticos con potencial hepatotóxico comprobado son los antimetabolitos, como la 6-mercaptopurina, la 6-tioguanina, el metotrexato y, con poca frecuencia, la dactinomicina.
Se observó enfermedad venoclusiva o síndrome de obstrucción sinusoidal (EVO/SOS), así como enfermedad colestática después de la administración de tiopurinas, en particular, 6-tioguanina.[60]
Se notificó fibrosis progresiva e hipertensión portal en un subgrupo de niños que presentaron EVO/SOS después del tratamiento con 6-tioguanina.[60]
Se observaron EVO/SOS agudos relacionados con la dosis y reversibles en niños tratados con dactinomicina por tumores sólidos infantiles.[61,62]
También se observaron EVO/SOS en el entorno de un trasplante después de regímenes de acondicionamiento que incluyeron ciclofosfamida o ICT, busulfano o ciclofosfamida, y carmustina, ciclofosfamida o etopósido.[63] Se especula que las dosis altas de ciclofosfamida, comunes en todos estos regímenes, son un posible factor causal.
Radioterapia
La hepatopatía aguda inducida por la radiación también provoca daño en las células endoteliales que es característico de EVO/SOS.[64]
En los adultos, todo el hígado tiene tolerancia de hasta 30 a 35 Gy con el fraccionamiento convencional, la prevalencia de enfermedad hepática inducida por radiación varía del 6 % al 66 %, según el volumen de hígado comprometido y la reserva hepática.[64,65]
La hepatopatía inducida por la radiación después del tratamiento contemporáneo es infrecuente en los sobrevivientes a largo plazo sin afecciones predisponentes como la hepatitis vírica o la sobrecarga de hierro.[66]
El riesgo de lesiones en niños aumenta con la dosis de radiación, el volumen hepático, la edad temprana en el momento del tratamiento, una hepatectomía parcial previa y el uso simultáneo de quimioterapia con radiomiméticos como la dactinomicina y la doxorrubicina.[67-70]
Los sobrevivientes sometidos a dosis de radiación de 40 Gy dirigida a, por lo menos, un tercio del volumen del hígado, dosis de 30 Gy o superiores dirigidas a todo el abdomen o al campo abdominal superior que incluye todo el hígado tienen el riesgo más alto de disfunción hepática.[42]
Se llevó a cabo una revisión exhaustiva del PENTEC para la creación de modelos que sirvan de base a las restricciones de dosis para la toxicidad hepática asociada a la radiación.[71]
En un modelo creado para calcular el riesgo de presentar SOS después de la radioterapia de todo el hígado, la dosis de radiación y la recepción de quimioterapia no alquilante fueron factores de riesgo significativos. La edad inferior a 20 años en el momento de la radioterapia fue casi significativa.
El riesgo previsto de SOS fue del 2 % con una dosis de radioterapia nula, del 6,1 % tras el tratamiento con 10 Gy y del 14,5 % tras el tratamiento con 20 Gy en todo el hígado.
Los pacientes con tumor de Wilms tratados con radioterapia en el flanco derecho (dosis estimada, 6,54 ± 2,50 Gy) presentaron una mayor tasa observada de SOS que los pacientes que recibieron radioterapia en el flanco izquierdo (dosis estimada, 2,16 ± 1,15 Gy).
Los datos fueron escasos con respecto a las tasas de lesión hepática tardía después de la radioterapia, lo que indica tasas bajas de toxicidad grave después del tratamiento para neoplasias malignas pediátricas comunes.
Trasplante de células madre hematopoyéticas
La disfunción hepática crónica en pacientes que se sometieron a un TCMH es de causa multifactorial, por ejemplo, sobrecarga de hierro, EICH crónica y hepatitis vírica.[72]
Los pacientes de EICH crónica en el aparato digestivo que presentan bilirrubina elevada tienen un pronóstico y una calidad de vida más precarios.[73]
Si bien la disfunción hepática crónica se observa en más de la mitad de los sobrevivientes de TCMH a largo plazo y la evolución de la enfermedad es lenta, es necesario un seguimiento continuo para establecer su efecto a largo plazo en la salud del sobreviviente.[74]
Factores infecciosos de riesgo de complicaciones hepatobiliares
En ocasiones, la hepatitis por los virus B y C complica el curso de tratamiento del cáncer infantil y produce una disfunción hepática crónica.
La incidencia de la hepatitis C relacionada con una transfusión en sobrevivientes de cáncer infantil oscila entre el 5 % y el 50 %, según la ubicación geográfica del centro notificador.[75-81]
La hepatitis crónica predispone a los sobrevivientes de cáncer infantil a cirrosis, hepatopatía en estadio terminal y carcinoma hepatocelular.[79,80]
La infección simultánea con ambos virus de hepatitis B y C combinados o con otros virus hepatotróficos acelera la progresión de la hepatopatía.[76,78]
Dado que la mayoría de los pacientes reciben algún tipo de hemoderivado durante el tratamiento de un cáncer infantil y a veces no son conscientes de sus antecedentes de transfusiones, se recomiendan los exámenes de detección según la fecha de diagnóstico o tratamiento.[82]
Todos los sobrevivientes de cáncer infantil que recibieron tratamiento antes de 1972 se deben someter a exámenes de detección de hepatitis B, además, todos los niños que recibieron transfusiones de sangre antes de 1993 se deben someter a exámenes de detección de hepatitis C y ser remitidos para analizar las opciones de tratamiento si los resultados de dichos exámenes son positivos.[82]
Atención posterior al tratamiento
Los sobrevivientes con disfunción hepática deberán recibir asesoramiento respecto a los métodos de reducción del riesgo para prevenir lesiones hepáticas.
Las recomendaciones estándar incluyen mantener un peso corporal saludable, abstenerse de consumir bebidas alcohólicas y vacunarse contra los virus de la hepatitis A y B.[42]
En los pacientes de hepatitis crónica, también se deberán analizar las precauciones para reducir la transmisión vírica en el hogar y a los contactos sexuales.
En el Cuadro 7 se resumen los efectos tardíos hepatobiliares y los exámenes médicos de detección correspondientes.
Cuadro 7. Efectos tardíos hepatobiliaresa
Tratamiento predisponente
Efectos hepáticos
Exámenes médicos de detección e intervenciones
ALT = alanina–aminotransferasa; AST = aspartato–aminotransferasa; TCMH = trasplante de células madre hematopoyéticas.
Exámenes de laboratorio: ALT, AST y concentraciones de bilirrubina
Ferritina en aquellos tratados con un TCMH
Mercaptopurina o tioguanina; TCMH
Enfermedad venoclusiva o síndrome de obstrucción sinusoidal
Hallazgos del examen: ictericia escleral, ictericia, ascitis, hepatomegalia y esplenomegalia
Exámenes de laboratorio: ALT, AST, concentración de bilirrubina y de plaquetas
Ferritina en aquellos tratados con un TCMH
Radioterapia con exposición del hígado o las vías biliares, y TCMH
Fibrosis hepática o cirrosis, hiperplasia focal nodular
Hallazgos del examen: ictericia, angiomas aracnoides, eritema palmar, xantomas, hepatomegalia y esplenomegalia
Exámenes de laboratorio: ALT, AST y concentraciones de bilirrubina
Ferritina en aquellos tratados con un TCMH
Tiempo de protrombina para evaluar la función de la síntesis hepática en pacientes con resultados anormales en los exámenes de detección de alteraciones hepáticas
Exámenes de detección de hepatitis vírica en pacientes con funcionamiento hepático anómalo persistente o en cualquier paciente que recibió una transfusión antes de 1993
Consulta de gastroenterología o hepatología en pacientes con disfunción hepática persistente
Vacunación contra hepatitis A y B en pacientes que carecen de inmunidad contra estos virus
Considerar una flebotomía y terapia con quelantes para una sobrecarga de hierro
Radioterapia con exposición del hígado o las vías biliares
Colelitiasis
Antecedentes: dolor cólico abdominal relacionado con el consumo de alimentos grasos y flatulencia excesiva
Hallazgos del examen: dolor a la palpación en el cuadrante superior derecho o en el epigastrio (episodio agudo)
Considerar una ecografía de la vesícula biliar para pacientes con dolor abdominal crónico
Páncreas
Se pensaba que el páncreas era relativamente resistente a la radiación debido a una escasez de información acerca de los efectos tardíos en el páncreas. Sin embargo, se sabe que los niños y los adultos jóvenes tratados con ITC o irradiación abdominal tienen un aumento de riesgo de resistencia a la insulina y diabetes mellitus.[83-85]
Si bien los corticoesteroides y la asparaginasa se relacionan con toxicidad aguda en el páncreas, no se han notificado secuelas tardías en el funcionamiento pancreático exocrino o endocrino para aquellos que sufren una lesión aguda.
Evidencia (riesgo de diabetes mellitus):
Se investigó, utilizando la cohorte SJLIFE, la prevalencia de prediabetes entre sobrevivientes de cáncer infantil, los factores de riesgo de progresión a diabetes y la relación entre la prediabetes y episodios cardiovasculares tardíos y la enfermedad renal crónica.[86]
Se evaluó la prevalencia de la prediabetes (definida como una concentración de glucosa plasmática en ayunas de 100–125 mg/dl o una hemoglobina A1c de 5,7–6,4 %) en 3529 sobrevivientes adultos de cáncer infantil. La prevalencia de la prediabetes en los sobrevivientes (mediana de edad, 30 años) fue del 29,2 %, y la prevalencia de la diabetes fue del 6,5 %.
Entre los 40 y 49 años de edad, la prevalencia de la prediabetes fue del 45,5 %, y en cada estrato de edad, los sobrevivientes tenían una prevalencia significativamente mayor de prediabetes y diabetes que los controles (P < 0,0025). Entre los 695 sobrevivientes con prediabetes que se sometieron a seguimiento longitudinal, el 10 % progresó a diabetes.
La exposición a la radiación en la cola del páncreas fue el único tratamiento que aumentó de manera significativa el riesgo de prediabetes, con independencia de la edad, el sexo, la raza, el IMC, la actividad física u otras exposiciones terapéuticas.
En comparación con los sobrevivientes con un control normal de la glucosa, ajustando por exposiciones relevantes al tratamiento, los sobrevivientes con prediabetes tuvieron un mayor riesgo de infarto de miocardio futuro (cociente de riesgos instantáneos [CRI], 2,4) y enfermedad renal crónica (CRI, 2,9), mientras que los sobrevivientes con diabetes también tuvieron un mayor riesgo de miocardiopatía futura (CRI, 3,8) y accidente cerebrovascular (CRI, 3,4).
Los investigadores del CCSS evaluaron el riesgo de diabetes mellitus entre 20 762 sobrevivientes a 5 años de cáncer infantil y 4853 hermanos.[87]
Los sobrevivientes que recibieron radiación abdominal (n = 4568) tuvieron una probabilidad 3 veces mayor de presentar diabetes que sus hermanos y 1,6 veces mayor que los sobrevivientes que no recibieron radiación abdominal.
Entre los sobrevivientes tratados con radioterapia abdominal, se identificaron factores de riesgo independientes para el origen de la diabetes mediante modelos multivariantes, entre estos, la edad más avanzada, el IMC más alto y el aumento de la dosis dirigida a la cola del páncreas.
También se identificó una interacción significativa entre la edad más joven (<10 años) en el momento del diagnóstico de cáncer y una dosis media más alta dirigida a la cola del páncreas.
Los investigadores del estudio SJLIFE evaluaron la prevalencia y los factores de riesgo de la diabetes mellitus entre 1044 adultos sobrevivientes de LLA infantil (media de edad, 34 años) quienes se evaluaron clínicamente más de 10 años después del tratamiento y 368 controles comunitarios (media de edad, 35 años).[88]
La tasa de prevalencia de la diabetes mellitus de tipo 2 fue del 7,5 % entre los sobrevivientes y del 3,8 % entre los controles.
Los factores de riesgo independiente para la diabetes entre los sobrevivientes incluyeron edad avanzada (OR, 1,05 por cada año adicional), IMC de 30 kg/m2 o mayor (OR, 7,4) y antecedentes de hiperglucemia inducida por fármacos durante el tratamiento (OR, 4,67).
En un estudio retrospectivo de cohortes sobre la base de las autonotificaciones de 2520 sobrevivientes a 5 años de cáncer infantil tratados en Francia y el Reino Unido, se investigó la relación entre la dosis de radiación dirigida al páncreas y el riesgo de un diagnóstico posterior de diabetes mellitus.[89]
Se validaron 65 casos de diabetes mellitus; el riesgo aumentó con la radioterapia dirigida a la cola del páncreas, donde se concentran los islotes de Langerhans. El riesgo aumentó con la administración de 20 a 29 Gy y luego se estabilizó. El RR calculado para 1 Gy fue de 1,61.
La dosis de radiación dirigida a otras partes del páncreas no tuvo un efecto considerable.
En comparación con los pacientes que no recibieron radioterapia, el RR de diabetes mellitus fue de 11,5 para los pacientes que recibieron más de 10 Gy dirigidos al páncreas.
Los niños menores de 2 años en el momento de la radioterapia fueron más sensibles que los pacientes de más edad (RR de 2,1 para 1 Gy en el grupo de edad más temprana vs. 1,4 para los pacientes mayores).
En los 511 pacientes que recibieron más de 10 Gy, la incidencia acumulada de diabetes mellitus fue del 16 %.
En otro estudio, se evaluó el riesgo de diabetes mellitus en 2264 sobrevivientes a 5 años de linfoma de Hodgkin (42 % menores de 25 años en el momento del diagnóstico) después de una mediana de seguimiento de 21,5 años.[90]
La incidencia acumulada de diabetes mellitus fue del 8,3 % (IC 95 %, 6,9–9,8 %) para toda la cohorte y del 14,2 % (IC 95 %, 10,7–18,3 %) para aquellos tratados con más de 36 Gy de radiación paraórtica.
Los sobrevivientes tratados con más de 36 Gy de radiación dirigida a los ganglios linfáticos paraórticos y el bazo tuvieron un riesgo de diabetes mellitus 2,3 veces mayor en comparación con aquellos que no recibieron radioterapia.
El riesgo de diabetes mellitus aumentó con dosis más altas dirigidas a la cola del páncreas.
Hölttä P, Alaluusua S, Saarinen-Pihkala UM, et al.: Agenesis and microdontia of permanent teeth as late adverse effects after stem cell transplantation in young children. Cancer 103 (1): 181-90, 2005. [PUBMED Abstract]
Hölttä P, Hovi L, Saarinen-Pihkala UM, et al.: Disturbed root development of permanent teeth after pediatric stem cell transplantation. Dental root development after SCT. Cancer 103 (7): 1484-93, 2005. [PUBMED Abstract]
Kaste SC, Goodman P, Leisenring W, et al.: Impact of radiation and chemotherapy on risk of dental abnormalities: a report from the Childhood Cancer Survivor Study. Cancer 115 (24): 5817-27, 2009. [PUBMED Abstract]
Paulino AC, Simon JH, Zhen W, et al.: Long-term effects in children treated with radiotherapy for head and neck rhabdomyosarcoma. Int J Radiat Oncol Biol Phys 48 (5): 1489-95, 2000. [PUBMED Abstract]
Maciel JC, de Castro CG, Brunetto AL, et al.: Oral health and dental anomalies in patients treated for leukemia in childhood and adolescence. Pediatr Blood Cancer 53 (3): 361-5, 2009. [PUBMED Abstract]
Hsieh SG, Hibbert S, Shaw P, et al.: Association of cyclophosphamide use with dental developmental defects and salivary gland dysfunction in recipients of childhood antineoplastic therapy. Cancer 117 (10): 2219-27, 2011. [PUBMED Abstract]
Dahllöf G: Oral and dental late effects after pediatric stem cell transplantation. Biol Blood Marrow Transplant 14 (1 Suppl 1): 81-3, 2008. [PUBMED Abstract]
Hölttä P, Alaluusua S, Saarinen-Pihkala UM, et al.: Long-term adverse effects on dentition in children with poor-risk neuroblastoma treated with high-dose chemotherapy and autologous stem cell transplantation with or without total body irradiation. Bone Marrow Transplant 29 (2): 121-7, 2002. [PUBMED Abstract]
Effinger KE, Migliorati CA, Hudson MM, et al.: Oral and dental late effects in survivors of childhood cancer: a Children's Oncology Group report. Support Care Cancer 22 (7): 2009-19, 2014. [PUBMED Abstract]
Kang CM, Hahn SM, Kim HS, et al.: Clinical Risk Factors Influencing Dental Developmental Disturbances in Childhood Cancer Survivors. Cancer Res Treat 50 (3): 926-935, 2018. [PUBMED Abstract]
Çetiner D, Çetiner S, Uraz A, et al.: Oral and dental alterations and growth disruption following chemotherapy in long-term survivors of childhood malignancies. Support Care Cancer 27 (5): 1891-1899, 2019. [PUBMED Abstract]
Patni T, Lee CT, Li Y, et al.: Factors for poor oral health in long-term childhood cancer survivors. BMC Oral Health 23 (1): 73, 2023. [PUBMED Abstract]
Thompson RF, Schneider RA, Albertini F, et al.: Dose to the developing dentition during therapeutic irradiation: organ at risk determination and clinical implications. Int J Radiat Oncol Biol Phys 86 (1): 108-13, 2013. [PUBMED Abstract]
Maguire A, Craft AW, Evans RG, et al.: The long-term effects of treatment on the dental condition of children surviving malignant disease. Cancer 60 (10): 2570-5, 1987. [PUBMED Abstract]
Paulino AC: Role of radiation therapy in parameningeal rhabdomyosarcoma. Cancer Invest 17 (3): 223-30, 1999. [PUBMED Abstract]
Kaste SC, Hopkins KP, Jones D, et al.: Dental abnormalities in children treated for acute lymphoblastic leukemia. Leukemia 11 (6): 792-6, 1997. [PUBMED Abstract]
Elzembely MM, Dahlberg AE, Pinto N, et al.: Late effects in high-risk neuroblastoma survivors treated with high-dose chemotherapy and stem cell rescue. Pediatr Blood Cancer 66 (1): e27421, 2019. [PUBMED Abstract]
Jensen SB, Pedersen AM, Vissink A, et al.: A systematic review of salivary gland hypofunction and xerostomia induced by cancer therapies: prevalence, severity and impact on quality of life. Support Care Cancer 18 (8): 1039-60, 2010. [PUBMED Abstract]
Jensen SB, Pedersen AM, Vissink A, et al.: A systematic review of salivary gland hypofunction and xerostomia induced by cancer therapies: management strategies and economic impact. Support Care Cancer 18 (8): 1061-79, 2010. [PUBMED Abstract]
Garming Legert K, Remberger M, Ringdèn O, et al.: Salivary secretion in children after fractionated or single-dose TBI. Bone Marrow Transplant 47 (3): 404-10, 2012. [PUBMED Abstract]
Bölling T, Weege J, Eich HT, et al.: Acute and late side effects to salivary glands and oral mucosa after head and neck radiotherapy in children and adolescents. Results of the "Registry for the evaluation of side effects after radiotherapy in childhood and adolescence". Head Neck 37 (8): 1137-41, 2015. [PUBMED Abstract]
Milgrom SA, van Luijk P, Pino R, et al.: Salivary and Dental Complications in Childhood Cancer Survivors Treated With Radiation Therapy to the Head and Neck: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 467-481, 2024. [PUBMED Abstract]
Dahllöf G, Wondimu B, Barr-Agholme M, et al.: Xerostomia in children and adolescents after stem cell transplantation conditioned with total body irradiation or busulfan. Oral Oncol 47 (9): 915-9, 2011. [PUBMED Abstract]
Garming-Legert K, Remberger M, Ringdén O, et al.: Long-term salivary function after conditioning with busulfan, fractionated or single-dose TBI. Oral Dis 17 (7): 670-6, 2011. [PUBMED Abstract]
Gevorgyan A, La Scala GC, Neligan PC, et al.: Radiation-induced craniofacial bone growth disturbances. J Craniofac Surg 18 (5): 1001-7, 2007. [PUBMED Abstract]
Sulaiman F, Huryn JM, Zlotolow IM: Dental extractions in the irradiated head and neck patient: a retrospective analysis of Memorial Sloan-Kettering Cancer Center protocols, criteria, and end results. J Oral Maxillofac Surg 61 (10): 1123-31, 2003. [PUBMED Abstract]
Vissink A, Jansma J, Spijkervet FK, et al.: Oral sequelae of head and neck radiotherapy. Crit Rev Oral Biol Med 14 (3): 199-212, 2003. [PUBMED Abstract]
Hong CH, Napeñas JJ, Hodgson BD, et al.: A systematic review of dental disease in patients undergoing cancer therapy. Support Care Cancer 18 (8): 1007-21, 2010. [PUBMED Abstract]
Goldsby R, Chen Y, Raber S, et al.: Survivors of childhood cancer have increased risk of gastrointestinal complications later in life. Gastroenterology 140 (5): 1464-71.e1, 2011. [PUBMED Abstract]
Bölling T, Willich N, Ernst I: Late effects of abdominal irradiation in children: a review of the literature. Anticancer Res 30 (1): 227-31, 2010. [PUBMED Abstract]
Churnratanakul S, Wirzba B, Lam T, et al.: Radiation and the small intestine. Future perspectives for preventive therapy. Dig Dis 8 (1): 45-60, 1990. [PUBMED Abstract]
Emami B, Lyman J, Brown A, et al.: Tolerance of normal tissue to therapeutic irradiation. Int J Radiat Oncol Biol Phys 21 (1): 109-22, 1991. [PUBMED Abstract]
Weil BR, Murphy AJ, Liu Q, et al.: Late Health Outcomes Among Survivors of Wilms Tumor Diagnosed Over Three Decades: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 41 (14): 2638-2650, 2023. [PUBMED Abstract]
Madenci AL, Fisher S, Diller LR, et al.: Intestinal Obstruction in Survivors of Childhood Cancer: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 33 (26): 2893-900, 2015. [PUBMED Abstract]
Madenci AL, Dieffenbach BV, Liu Q, et al.: Late-onset anorectal disease and psychosocial impact in survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 125 (21): 3873-3881, 2019. [PUBMED Abstract]
Heyn R, Raney RB, Hays DM, et al.: Late effects of therapy in patients with paratesticular rhabdomyosarcoma. Intergroup Rhabdomyosarcoma Study Committee. J Clin Oncol 10 (4): 614-23, 1992. [PUBMED Abstract]
Hughes LL, Baruzzi MJ, Ribeiro RC, et al.: Paratesticular rhabdomyosarcoma: delayed effects of multimodality therapy and implications for current management. Cancer 73 (2): 476-82, 1994. [PUBMED Abstract]
Raney B, Heyn R, Hays DM, et al.: Sequelae of treatment in 109 patients followed for 5 to 15 years after diagnosis of sarcoma of the bladder and prostate. A report from the Intergroup Rhabdomyosarcoma Study Committee. Cancer 71 (7): 2387-94, 1993. [PUBMED Abstract]
Asdahl PH, Oeffinger KC, Albieri V, et al.: Esophageal disease among childhood cancer survivors-A report from the Childhood Cancer Survivors Study. Pediatr Blood Cancer 68 (8): e29043, 2021. [PUBMED Abstract]
Mulder RL, van Dalen EC, Van den Hof M, et al.: Hepatic late adverse effects after antineoplastic treatment for childhood cancer. Cochrane Database Syst Rev (7): CD008205, 2011. [PUBMED Abstract]
Castellino S, Muir A, Shah A, et al.: Hepato-biliary late effects in survivors of childhood and adolescent cancer: a report from the Children's Oncology Group. Pediatr Blood Cancer 54 (5): 663-9, 2010. [PUBMED Abstract]
Mulder RL, Kremer LC, Koot BG, et al.: Surveillance of hepatic late adverse effects in a large cohort of long-term survivors of childhood cancer: prevalence and risk factors. Eur J Cancer 49 (1): 185-93, 2013. [PUBMED Abstract]
Green DM, Wang M, Krasin MJ, et al.: Serum Alanine Aminotransferase Elevations in Survivors of Childhood Cancer: A Report From the St. Jude Lifetime Cohort Study. Hepatology 69 (1): 94-106, 2019. [PUBMED Abstract]
Castellino SM, Hudson MM: Late gastrointestinal and hepatic effects. In: Schwartz
CL, Hobbie WL,
Constine LS, et al., eds.: Survivors of Childhood and Adolescent Cancer: A Multidisciplinary Approach. 3rd ed. Springer International Publishing, 2015, pp 229-52.
Mahmoud H, Schell M, Pui CH: Cholelithiasis after treatment for childhood cancer. Cancer 67 (5): 1439-42, 1991. [PUBMED Abstract]
Safford SD, Safford KM, Martin P, et al.: Management of cholelithiasis in pediatric patients who undergo bone marrow transplantation. J Pediatr Surg 36 (1): 86-90, 2001. [PUBMED Abstract]
Dieffenbach BV, Li N, Madenci AL, et al.: Incidence of and risk factors for late cholecystectomy in survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Eur J Cancer 133: 4-13, 2020. [PUBMED Abstract]
Sudour H, Mainard L, Baumann C, et al.: Focal nodular hyperplasia of the liver following hematopoietic SCT. Bone Marrow Transplant 43 (2): 127-32, 2009. [PUBMED Abstract]
Lee MH, Yoo SY, Kim JH, et al.: Hypervascular hepatic nodules in childhood cancer survivors: clinical and imaging features. Clin Imaging 36 (4): 301-7, 2012 Jul-Aug. [PUBMED Abstract]
Wanless IR: Micronodular transformation (nodular regenerative hyperplasia) of the liver: a report of 64 cases among 2,500 autopsies and a new classification of benign hepatocellular nodules. Hepatology 11 (5): 787-97, 1990. [PUBMED Abstract]
Brisse H, Servois V, Bouche B, et al.: Hepatic regenerating nodules: a mimic of recurrent cancer in children. Pediatr Radiol 30 (6): 386-93, 2000. [PUBMED Abstract]
Chu WC, Roebuck DJ: Nodular regenerative hyperplasia of the liver simulating metastases following treatment for bilateral Wilms tumor. Med Pediatr Oncol 41 (1): 85-7, 2003. [PUBMED Abstract]
Halonen P, Mattila J, Ruuska T, et al.: Liver histology after current intensified therapy for childhood acute lymphoblastic leukemia: microvesicular fatty change and siderosis are the main findings. Med Pediatr Oncol 40 (3): 148-54, 2003. [PUBMED Abstract]
Tomita Y, Ishiguro H, Yasuda Y, et al.: High incidence of fatty liver and insulin resistance in long-term adult survivors of childhood SCT. Bone Marrow Transplant 46 (3): 416-25, 2011. [PUBMED Abstract]
Ruccione KS, Wood JC, Sposto R, et al.: Characterization of transfusion-derived iron deposition in childhood cancer survivors. Cancer Epidemiol Biomarkers Prev 23 (9): 1913-9, 2014. [PUBMED Abstract]
Vag T, Kentouche K, Krumbein I, et al.: Noninvasive measurement of liver iron concentration at MRI in children with acute leukemia: initial results. Pediatr Radiol 41 (8): 980-4, 2011. [PUBMED Abstract]
Baskin-Miller J, Carson S, Jaffray J, et al.: Transfusional hemosiderosis in childhood cancer patients and survivors. Pediatr Blood Cancer 71 (10): e31220, 2024. [PUBMED Abstract]
Schempp A, Lee J, Kearney S, et al.: Iron Overload in Survivors of Childhood Cancer. J Pediatr Hematol Oncol 38 (1): 27-31, 2016. [PUBMED Abstract]
Broxson EH, Dole M, Wong R, et al.: Portal hypertension develops in a subset of children with standard risk acute lymphoblastic leukemia treated with oral 6-thioguanine during maintenance therapy. Pediatr Blood Cancer 44 (3): 226-31, 2005. [PUBMED Abstract]
Green DM, Norkool P, Breslow NE, et al.: Severe hepatic toxicity after treatment with vincristine and dactinomycin using single-dose or divided-dose schedules: a report from the National Wilms' Tumor Study. J Clin Oncol 8 (9): 1525-30, 1990. [PUBMED Abstract]
Sulis ML, Bessmertny O, Granowetter L, et al.: Veno-occlusive disease in pediatric patients receiving actinomycin D and vincristine only for the treatment of rhabdomyosarcoma. J Pediatr Hematol Oncol 26 (12): 843-6, 2004. [PUBMED Abstract]
McDonald GB: Hepatobiliary complications of hematopoietic cell transplantation, 40 years on. Hepatology 51 (4): 1450-60, 2010. [PUBMED Abstract]
Dawson LA, Ten Haken RK: Partial volume tolerance of the liver to radiation. Semin Radiat Oncol 15 (4): 279-83, 2005. [PUBMED Abstract]
Milano MT, Constine LS, Okunieff P: Normal tissue tolerance dose metrics for radiation therapy of major organs. Semin Radiat Oncol 17 (2): 131-40, 2007. [PUBMED Abstract]
Pan CC, Kavanagh BD, Dawson LA, et al.: Radiation-associated liver injury. Int J Radiat Oncol Biol Phys 76 (3 Suppl): S94-100, 2010. [PUBMED Abstract]
Bhanot P, Cushing B, Philippart A, et al.: Hepatic irradiation and adriamycin cardiotoxicity. J Pediatr 95 (4): 561-3, 1979. [PUBMED Abstract]
Flentje M, Weirich A, Pötter R, et al.: Hepatotoxicity in irradiated nephroblastoma patients during postoperative treatment according to SIOP9/GPOH. Radiother Oncol 31 (3): 222-8, 1994. [PUBMED Abstract]
Kun LE, Camitta BM: Hepatopathy following irradiation and adriamycin. Cancer 42 (1): 81-4, 1978. [PUBMED Abstract]
Tefft M: Radiation related toxicities in National Wilms' Tumor Study Number 1. Int J Radiat Oncol Biol Phys 2 (5-6): 455-63, 1977 May-Jun. [PUBMED Abstract]
Hall MD, Howell RM, Jackson A, et al.: Liver Late Effects in Childhood Cancer Survivors Treated With Radiation Therapy: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 575-587, 2024. [PUBMED Abstract]
Pidala J, Chai X, Kurland BF, et al.: Analysis of gastrointestinal and hepatic chronic graft-versus-host [corrected] disease manifestations on major outcomes: a chronic graft-versus-host [corrected] disease consortium study. Biol Blood Marrow Transplant 19 (5): 784-91, 2013. [PUBMED Abstract]
Tomás JF, Pinilla I, García-Buey ML, et al.: Long-term liver dysfunction after allogeneic bone marrow transplantation: clinical features and course in 61 patients. Bone Marrow Transplant 26 (6): 649-55, 2000. [PUBMED Abstract]
Aricò M, Maggiore G, Silini E, et al.: Hepatitis C virus infection in children treated for acute lymphoblastic leukemia. Blood 84 (9): 2919-22, 1994. [PUBMED Abstract]
Castellino S, Lensing S, Riely C, et al.: The epidemiology of chronic hepatitis C infection in survivors of childhood cancer: an update of the St Jude Children's Research Hospital hepatitis C seropositive cohort. Blood 103 (7): 2460-6, 2004. [PUBMED Abstract]
Cesaro S, Petris MG, Rossetti F, et al.: Chronic hepatitis C virus infection after treatment for pediatric malignancy. Blood 90 (3): 1315-20, 1997. [PUBMED Abstract]
Fink FM, Höcker-Schulz S, Mor W, et al.: Association of hepatitis C virus infection with chronic liver disease in paediatric cancer patients. Eur J Pediatr 152 (6): 490-2, 1993. [PUBMED Abstract]
Locasciulli A, Testa M, Pontisso P, et al.: Hepatitis C virus genotypes and liver disease in patients undergoing allogeneic bone marrow transplantation. Bone Marrow Transplant 19 (3): 237-40, 1997. [PUBMED Abstract]
Locasciulli A, Testa M, Pontisso P, et al.: Prevalence and natural history of hepatitis C infection in patients cured of childhood leukemia. Blood 90 (11): 4628-33, 1997. [PUBMED Abstract]
Paul IM, Sanders J, Ruggiero F, et al.: Chronic hepatitis C virus infections in leukemia survivors: prevalence, viral load, and severity of liver disease. Blood 93 (11): 3672-7, 1999. [PUBMED Abstract]
Lansdale M, Castellino S, Marina N, et al.: Knowledge of hepatitis C virus screening in long-term pediatric cancer survivors: a report from the Childhood Cancer Survivor Study. Cancer 116 (4): 974-82, 2010. [PUBMED Abstract]
van Waas M, Neggers SJ, Raat H, et al.: Abdominal radiotherapy: a major determinant of metabolic syndrome in nephroblastoma and neuroblastoma survivors. PLoS One 7 (12): e52237, 2012. [PUBMED Abstract]
Neville KA, Cohn RJ, Steinbeck KS, et al.: Hyperinsulinemia, impaired glucose tolerance, and diabetes mellitus in survivors of childhood cancer: prevalence and risk factors. J Clin Endocrinol Metab 91 (11): 4401-7, 2006. [PUBMED Abstract]
Baker KS, Ness KK, Steinberger J, et al.: Diabetes, hypertension, and cardiovascular events in survivors of hematopoietic cell transplantation: a report from the bone marrow transplantation survivor study. Blood 109 (4): 1765-72, 2007. [PUBMED Abstract]
Dixon SB, Wang F, Lu L, et al.: Prediabetes and Associated Risk of Cardiovascular Events and Chronic Kidney Disease Among Adult Survivors of Childhood Cancer in the St Jude Lifetime Cohort. J Clin Oncol 42 (9): 1031-1043, 2024. [PUBMED Abstract]
Friedman DN, Moskowitz CS, Hilden P, et al.: Radiation Dose and Volume to the Pancreas and Subsequent Risk of Diabetes Mellitus: A Report from the Childhood Cancer Survivor Study. J Natl Cancer Inst 112 (5): 525-532, 2020. [PUBMED Abstract]
Williams HE, Howell CR, Chemaitilly W, et al.: Diabetes mellitus among adult survivors of childhood acute lymphoblastic leukemia: A report from the St. Jude Lifetime Cohort Study. Cancer 126 (4): 870-878, 2020. [PUBMED Abstract]
de Vathaire F, El-Fayech C, Ben Ayed FF, et al.: Radiation dose to the pancreas and risk of diabetes mellitus in childhood cancer survivors: a retrospective cohort study. Lancet Oncol 13 (10): 1002-10, 2012. [PUBMED Abstract]
van Nimwegen FA, Schaapveld M, Janus CP, et al.: Risk of diabetes mellitus in long-term survivors of Hodgkin lymphoma. J Clin Oncol 32 (29): 3257-63, 2014. [PUBMED Abstract]
Efectos tardíos en el sistema endocrino
La disfunción endocrina es común entre los sobrevivientes de cáncer infantil, en especial en aquellos que reciben tratamiento mediante cirugía o radioterapia que comprometieron órganos productores de hormonas, así como aquellos que recibieron quimioterapia con alquilantes.
La prevalencia de trastornos endocrinos específicos está afectada por los siguientes factores:[1-4]
Factores del paciente (por ejemplo, edad en el momento del tratamiento y sexo).
Ubicación del tumor (por ejemplo, supraselar y no supraselar).[5]
Factores del tratamiento (por ejemplo, dosis de radiación y volumen tratado).
Tiempo transcurrido desde la exposición a la radiación (por lo general aumenta cuanto mayor es el tiempo transcurrido desde la exposición a la radiación [consultar la Figura 6]).[1]
Los efectos endocrinológicos tardíos se clasifican de manera amplia como aquellos originados en alguna lesión hipotalámica o hipofisaria, o por compromiso glandular periférico.[1-4] Los primeros son más comunes después del tratamiento de tumores en el sistema nervioso central (SNC), para los cuales se notificó una prevalencia del 24,8 % en un estudio de cohorte nacional de 718 sobrevivientes que vivieron más de 2 años y que tenían compromiso de todo el sistema hipotalamohipofisario.[3]
En las siguientes secciones se resume la investigación sobre las características clínicas de los sobrevivientes con riesgo de disfunción endocrina que incide en el funcionamiento hipofisario, tiroideo, suprarrenal y gonadal.
Glándula tiroidea
La disfunción tiroidea es un efecto tardío frecuente de los campos de radioterapia que abarcan la glándula tiroidea. Este tipo de radioterapia se usa para tratar el linfoma de Hodgkin, los tumores de encéfalo, los sarcomas de cabeza y cuello y la leucemia linfoblástica aguda (LLA).
Se cuenta con evidencia científica considerable que vincula la exposición a la radiación con anomalías tiroideas; sin embargo, la prevalencia de afecciones específicas varía mucho porque los estudios están limitados debido al sesgo de selección y participación de la cohorte, la heterogeneidad en el abordaje de tratamiento con radiación, el tiempo transcurrido desde la exposición a la radiación y el método de confirmación (por ejemplo, autonotificación vs. evaluación clínica o evaluación diagnóstica con imágenes).
Las anomalías tiroideas observadas en exceso en sobrevivientes de cáncer infantil son las siguientes:
Hipotiroidismo primario.
Hipertiroidismo.
Bocio.
Nódulos.
Hipotiroidismo
Factores de riesgo
Radioterapia tiroidea. Se notificó un aumento del riesgo de hipotiroidismo en niños sobrevivientes de cáncer tratados con radiación dirigida a la cabeza y el cuello con exposición de la glándula tiroidea, en particular, en sobrevivientes de linfoma de Hodgkin.[1-4] Los sobrevivientes de tumores de cabeza y cuello que recibieron radioterapia con posible exposición de la región hipotalamohipofisaria y de la glándula tiroides, quizás presenten hipotiroidismo como resultado de la deficiencia de la hormona liberadora de tirotropina (TRH) o de la hormona estimulante de la tiroides (TSH) (hipotiroidismo de origen central), disfunción de la glándula tiroides (hipotiroidismo primario), o una combinación de origen central y primario.
Yodo I 131-metayodobencilguanidina (131I-MIBG).
131I-MIBG terapéutico. Se notificaron trastornos tiroideos, incluso hipotiroidismo primario, en niños con neuroblastoma tratados con 131I-MIBG, a pesar de la protección de la tiroides con yoduro de potasio, perclorato o la combinación de yoduro potásico, tiroxina (T4) y tiamazol. Sin embargo, estos informes presentan un número pequeño de pacientes que tienen otros factores de riesgo relacionados con el tratamiento de trastornos tiroideos, lo que limita la capacidad de identificar un efecto independiente del 131I-MIBG.[6] Se diagnosticaron trastornos tiroideos en el 81 % de los sobrevivientes 15 años después del tratamiento con 131I-MIBG.[7]
131I-MIBG diagnóstico. No se notificaron anomalías tiroideas de inicio tardío en sobrevivientes de cáncer infantil cuyo estado de la enfermedad se controló mediante 131I-MIBG o 123I-MIBG.[8,9]
En un estudio retrospectivo de cohortes (n = 48) se evaluó la prevalencia de la disfunción tiroidea en sobrevivientes de un tumor neuroblástico que recibieron 123I-MIBG con fines diagnósticos. Todos los pacientes recibieron profilaxis tiroidea (yoduro de potasio o combinación de yoduro potásico, tiamazol y T4) durante la exposición a 123I-MIBG. Después de una mediana de seguimiento de 6,6 años, la función tiroidea fue normal en 46 de los 48 sobrevivientes. De los sobrevivientes, 2 presentaron disfunción tiroidea leve.[10]
Tiroidectomía. Como se observó en la población general sin cáncer, la resección quirúrgica parcial u, obviamente, subtotal de la tiroides es un factor de riesgo de hipotiroidismo subsiguiente.[11-13]
Evidencia (prevalencia y factores de riesgo de hipotiroidismo):
El German Group of Paediatric Radiation Oncology informó sobre 1086 pacientes tratados en 62 centros; entre ellos, 404 pacientes (mediana de edad, 10,9 años) que recibieron radioterapia dirigida a la tiroides o la hipófisis.[14] Se dispuso de información de seguimiento para 264 pacientes (60,9 %; mediana de seguimiento, 40 meses); 60 pacientes (22,7 %) mostraron valores patológicos.
En comparación con los pacientes que recibieron tratamiento con irradiación craneal profiláctica (mediana de dosis, 12 Gy), los pacientes tratados con dosis de radiación de 15 a 25 Gy dirigida a la glándula tiroidea tuvieron un cociente de riesgos instantáneos (CRI) de 3072 (P = 0,002) de presentar valores tiroideos patológicos en la sangre.
Los pacientes tratados con más de 25 Gy de radiación dirigida a la glándula tiroidea tuvieron un CRI fue de 3,769 (P = 0,009). Los pacientes sometidos a irradiación craneoespinal (ICE) tuvieron un CRI de 5,674 (P < 0,001).
La incidencia acumulada de terapia de reemplazo de la hormona tiroidea no difirió entre los subgrupos definidos.
El Childhood Cancer Survivor Study (CCSS) investigó la prevalencia del hipotiroidismo autonotificado evaluado mediante cuestionarios seriales en 12 015 sobrevivientes. Se observaron 1193 casos de hipotiroidismo, 777 (65 %) de los cuales se presentaron 5 años o más después del diagnóstico de cáncer.[15]
La prevalencia a los 5 años del diagnóstico de cáncer y la incidencia a lo largo de 30 años después del diagnóstico de cáncer fue más alta en los sobrevivientes a 5 años de linfoma de Hodgkin (32,3 %) y tumores del SNC (17,7 %).
La incidencia guardó una relación significativa con la dosis de radiación dirigida a la tiroides y la hipófisis. Los efectos combinados de las dosis dirigidas a la tiroides, el hipotálamo y la hipófisis son menos que aditivas cuando las dosis hipofisarias son mayores de 16 Gy.
Los riesgos de la radiación fueron más elevados en los varones que en las mujeres y guardaron relación inversa con la edad en el momento de la exposición y el tiempo transcurrido desde la exposición, pero permanecieron elevados más de 25 años después de la exposición.
Ciertos tipos de quimioterapia se relacionaron en forma significativa con el riesgo: bleomicina (razón de tasas, 3,4) y los fármacos alquilantes lomustina (razón de tasas, 3,0) y ciclofosfamida (cociente de tasas, 1,3). El riesgo de quimioterapia más importante ocurrió entre sobrevivientes de cáncer del SNC. Se observó una respuesta significativa a la dosis de lomustina (P < 0,01).
En el CCSS, se evaluó la enfermedad tiroidea en una cohorte de sobrevivientes de linfoma de Hodgkin infantil tratados entre 1970 y 1986 mediante un cuestionario autonotificado.[16]
Entre 1791 sobrevivientes que recibieron seguimiento durante una mediana de 14 años, el 34 % notificó que había sido diagnosticado con al menos una anomalía tiroidea.
Para el hipotiroidismo, hubo una respuesta bien definida según la dosis y el riesgo a 20 años fue como sigue:
Un 20 % para quienes recibieron menos de 35 Gy de radiación dirigida a la glándula tiroidea.
Un 30 % para quienes recibieron de 35 a 44,9 Gy de radiación dirigida a la glándula tiroidea.
Un 50 % para quienes recibieron más de 45 Gy de radiación dirigida a la glándula tiroidea.
En comparación con un grupo de control de hermanos, el riesgo relativo (RR) de hipotiroidismo fue de 17,1.
El tiempo transcurrido desde el diagnóstico fue un factor de riesgo de hipotiroidismo y el riesgo aumentó en los primeros 3 a 5 años posteriores al diagnóstico.
Las mujeres tuvieron un aumento de riesgo de hipotiroidismo.
En un estudio de seguimiento del CCSS se compararon los datos autonotificados de 14 290 sobrevivientes con los datos de 4031 hermanos del grupo de control.[2]
El RR fue de 3,8 para el hipotiroidismo y siguió siendo significativamente más alto en los sobrevivientes, en comparación con los controles, incluso sin administración de radioterapia dirigida a la tiroides o la hipófisis.
Estos resultados indican la necesidad de poner en práctica estrategias de seguimiento individualizadas y continuas a largo plazo para los sobrevivientes de cáncer infantil.
Las mejoras continuas en la precisión de la radioterapia son prometedoras para disminuir la dosis de radioterapia recibida por la tiroides en un subconjunto de pacientes, como se demostró en un estudio de 189 niños y adultos jóvenes (edad <26 años) con tumor de encéfalo tratados con radioterapia de protones.[17]
Al cabo de una mediana de seguimiento de 4,4 años, la incidencia acumulada de hipotiroidismo primario fue del 3 % después de la ICE y del 1,6 % en general, la cual es mucho más baja que en informes previos del 56 % al 65 % de incidencia después de la ICE con fotones.
Entre 118 sobrevivientes de meduloblastoma (mediana de seguimiento, 5,6 años), la incidencia acumulada estimada de hipotiroidismo fue del 35,8 % (intervalo de confianza [IC] 95 %, 26,7–47,1 %) a los 5 años de la radioterapia.[18]
La mediana de tiempo desde el final de la radioterapia hasta la presentación de hipotiroidismo fue de 2,1 años (intervalo, 0,1–7,1 años) después de la radioterapia de protones y de 2,9 años (intervalo, 0,8–7,2 años) después de la radioterapia con fotones.
La tasa de hipotiroidismo (CRI, 2,36; IC 95 %, 1,11–5,02) fue más alta en los pacientes a los que se trató con radioterapia de fotones, en comparación con los que recibieron tratamiento con radioterapia de protones.
Las diferencias en el riesgo de hipotiroidismo según la modalidad de radiación se atribuyeron en gran medida a tasas más altas de hipotiroidismo primario después de la radioterapia con fotones (CRI, 4,61; IC 95 %, 1,20–17,66).
Los investigadores del estudio de cohorte St. Jude Life (SJLIFE) evaluaron la prevalencia y los factores de riesgo del hipotiroidismo primario, así como su relación con las afecciones crónicas y la calidad de vida de 2965 sobrevivientes de cáncer infantil (mediana de edad, 30,9 años; mediana de tiempo después del diagnóstico, 22,3 años).[13]
El hipotiroidismo primario fue prevalente en el 14,7 % de los sobrevivientes. Fue más frecuente en las mujeres (oportunidad relativa [OR], 1,06; IC 95 %, 1,03–1,08) y en los sobrevivientes diagnosticados con más de 15 años (vs. edad <5 años). Fue menos común en los sobrevivientes que no eran blancos (OR, 0,96; IC 95 %, 0,93–0,99).
La radioterapia que potencialmente expone la glándula tiroides se relacionó con riesgo de hipotiroidismo relacionado con la dosis.
La fragilidad (OR, 1,54; IC 95 %, 1,05–2,26), la dislipidemia (OR, 1,52; IC 95 %, 1,14–2,04) y el deterioro de la calidad de vida física (OR, 1,66; IC 95 %, 1,12–2,48) se relacionaron de manera significativa con el hipotiroidismo primario.
El consorcio de investigación Pediatric Normal Tissue Effects in the Clinic (PENTEC) realizó una revisión sistemática del hipotiroidismo primario relacionado con la radiación en niños menores de 21 años entre 1970 y 2017.[19]
Las tasas previstas de hipotiroidismo no compensado (clínico) fueron, a dosis medias de 10 Gy, 20 Gy y 30 Gy, del 4 %, 7 % y 13 %, respectivamente. Las tasas de hipotiroidismo compensado (subclínico) fueron del 12 %, 25 % y 44 %, respectivamente.
El sexo femenino (RR, 1,7; P < 0,0001) y la edad menor de 15 años en el momento de la radioterapia (RR, 1,3; P < 0,005) se relacionaron con un riesgo más alto de hipotiroidismo.
Después de una dosis tiroidea media de 20 Gy, las tasas previstas de hipotiroidismo fueron del 13 % para los hombres menores de 14 años y más del 29 % para las mujeres mayores de 15 años en el momento de la radioterapia.
Cuadro 8. Riesgos de hipotiroidismo (compensado o no compensado) después de la radioterapia para neoplasias malignas infantiles, agrupados por dosis tiroidea media, edad y sexoa
bCualquier hipotiroidismo (es decir, compensado o no compensado).
cLa edad de 14 a 15 años se utilizó como límite, porque en los 2 estudios que analizaron la edad se utilizaron diferentes puntos de corte. Es de suponer que los riesgos de hipotiroidismo en pacientes irradiados entre los 14 y los 15 años de edad serían intermedios con respecto a los que se muestran para las edades <14 y >15 años.
10 Gy
10 %
6 %
14 %
8 %
20 Gy
22 %
13 %
29 %
17 %
30 Gy
39 %
23 %
53 %
31 %
40 Gy
59 %
35 %
79%
47%
El consorcio de investigación del PENTEC realizó una revisión sistemática del hipotiroidismo relacionado con la radiación.[20]
Entre 7 cohortes (250 pacientes), la dosis de 39 Gy se relacionó con un riesgo de hipotiroidismo del 50 % (D50) (IC 95 %, 34,1–53,2).
En los niños que recibieron una dosis media de 22 Gy en fracciones de 2 Gy, el riesgo fue del 20 %.
Cuadro clínico inicial
La mayoría de los niños tratados con radioterapia presentan hipotiroidismo en los primeros 2 a 5 años posteriores al tratamiento, pero a veces se presentan casos nuevos más tarde.
Los informes sobre la disfunción tiroidea difieren según la dosis de radiación, la duración del seguimiento y los criterios bioquímicos utilizados para establecer el diagnóstico.[21]
Las anomalías que se notifican con mayor frecuencia son las siguientes:
Elevación de la TSH.
Disminución de la tiroxina (T4).
Elevación de la TSH y disminución de la T4.
El hipotiroidismo central es producto de la exposición a la radiación del sistema hipotalamohipofisario; por lo general son bajas las concentraciones de T4 libre y TSH.
El hipotiroidismo compensado incluye una elevación de la TSH con una T4 normal y es asintomático. La evolución natural no está clara, pero la mayoría de los endocrinólogos respaldan su tratamiento.
El hipotiroidismo no compensado incluye una elevación de la TSH y una disminución de la T4.
El reemplazo de la hormona tiroidea es beneficioso para corregir la anomalía metabólica y ofrece beneficios clínicos para el funcionamiento cardiovascular, gastrointestinal y neurocognitivo.
Hipertiroidismo
Si bien el hipertiroidismo es menos común que el hipotiroidismo, los sobrevivientes de cáncer infantil también se enfrentan a un riesgo mayor de hipertiroidismo.[2,16,22,23]
Evidencia (prevalencia y factores de riesgo de hipertiroidismo):
Los investigadores del CCSS evaluaron la prevalencia de la enfermedad tiroidea en 1791 sobrevivientes de linfoma de Hodgkin infantil tratados entre 1970 y 1986, a los que se les realizó seguimiento durante una mediana de 14 años.[16]
Entre los sobrevivientes, el 5 % notificó hipertiroidismo, lo cual fue 8 veces superior a la incidencia notificada por los controles.
La dosis de radiación dirigida a la tiroides de 35 Gy o más fue el único factor de riesgo identificado para el hipertiroidismo.
En otro estudio del CCSS se evaluó el riesgo de hipertiroidismo en relación con la dosis de radiación terapéutica accidental en la glándula tiroidea y la hipófisis.[22]
El hipertiroidismo fue autonotificado por 179 sobrevivientes, con 148 casos diagnosticados 5 o más años después del diagnóstico de cáncer.
La proporción acumulada de sobrevivientes con hipertiroidismo a los 30 años después del diagnóstico de cáncer fue del 2,5 % (IC 95 %, 2,0–2,9 %).
La radiación tiroidea incrementó el riesgo de hipertiroidismo con evidencia científica de una relación lineal entre la dosis de radiación tiroidea y la respuesta, para un intervalo de la dosis de 0 a 63 Gy.
La dosis de radiación dirigida a la hipófisis y la quimioterapia no guardaron ninguna relación significativa con el hipertiroidismo.
El riesgo de hipertiroidismo asociado con la radiación siguió siendo elevado durante más de 25 años después de la exposición.
Nódulos tiroideos
La manifestación clínica de una neoplasia tiroidea en los sobrevivientes de cáncer infantil varía de nódulos solos, pequeños y asintomáticos, a bocios intratorácicos que oprimen las estructuras adyacentes.
Los siguientes factores se vinculan con un aumento del riesgo de nódulos tiroideos:
Dosis de radiación, tiempo transcurrido desde el diagnóstico y sexo femenino.
Cualquier campo de radiación que incluya la tiroides se relaciona con un exceso de riesgo de neoplasias tiroideas benignas (por lo general, adenomas) o malignas (con mayor frecuencia, carcinoma papilar diferenciado).[2,16,24-27]
En un estudio del CCSS en el que participaron sobrevivientes de linfoma de Hodgkin, se identificó el tiempo transcurrido desde el diagnóstico, el sexo femenino y la dosis de radiación de 25 Gy o más como factores de riesgo significativos para el desarrollo de nódulos tiroideos.[16]
En una cohorte de 3254 sobrevivientes de cáncer infantil a 2 años, tratados antes de 1986 y controlados por 25 años, el riesgo de adenoma tiroideo aumentó con el tamaño de la dosis de radiación dirigida a la tiroides durante el tratamiento del cáncer infantil y se estabilizó en dosis que excedieron los 10 Gy.[25]
Los investigadores del CCSS realizaron un estudio de casos y controles anidado para evaluar la magnitud del riesgo de cáncer de tiroides en relación con el intervalo de dosis de radiación terapéutica de los cánceres infantiles. El riesgo de cáncer de tiroides aumentó con dosis de radiación de 20 a 29 Gy (OR, 9,8; IC 95 %, 3,2–34,8), pero disminuyó con dosis mayores de 30 Gy, lo cual es congruente con un efecto citolítico.[27]
Edad en el momento de la radioterapia.
En la misma cohorte de 3254 sobrevivientes de cáncer infantil a 2 años, el riesgo de adenoma tiroideo por unidad de dosis de radiación dirigida a la tiroides fue más alto cuando la radioterapia se administró antes de los 5 años de edad; el riesgo también fue más alto en las personas menores de 40 años en el momento del estudio.[25]
Una edad menor en el momento de la radioterapia también se relacionó con un exceso de riesgo de carcinoma de tiroides.[24-27]
Exposición al 131I-MIBG.
Durante la niñez y la adolescencia hay un mayor riesgo de nódulos tiroideos y, posiblemente, cáncer de tiroides en pacientes expuestos a 131l-MIBG.
Los profesionales clínicos que atienden a los sobrevivientes tratados con 131I-MIBG deben ser conscientes de los riesgos de presentar disfunción tiroidea, nódulos tiroideos o cáncer de tiroides.[28]
Quimioterapia.
También se observó un aumento de riesgo de nódulos tiroideos y cáncer de tiroides en relación con la quimioterapia, de manera independiente de la exposición a la radiación.[2,24,25]
En un estudio combinado de 2 cohortes de 16 757 sobrevivientes en el que se incluyó a 187 pacientes con cáncer de tiroides secundario, los tratamientos con alquilantes, antraciclinas o bleomicina se relacionaron con un aumento significativo de riesgo de cáncer de tiroides en personas que no se habían expuesto a radioterapia.[29]
En el CCSS, la razón de tasas de presentar cáncer de tiroides fue de 2,5 (P < 0,01) en los sobrevivientes que no recibieron radiación dirigida a la tiroides, en comparación con los hermanos del grupo de control.[2]
Las áreas de investigación activa incluyen la definición de la función precisa de la exposición a la quimioterapia y la formulación de modelos pronósticos del riesgo de cáncer de tiroides para los sobrevivientes de cáncer infantil, a partir de los factores de riesgo demográficos y del tratamiento.[30]
Exámenes de detección del cáncer de tiroides
En varios estudios se demostró la superioridad de la ecografía en relación con el examen clínico para detectar nódulos tiroideos y cánceres de tiroides, y que determina las características ultrasonográficas de nódulos que tienen más probabilidades de ser malignos.[31-33]
Sin embargo, los exámenes de detección primaria de una neoplasia tiroidea (más allá del examen físico con palpación de la tiroides) continúan siendo polémicos por la carencia de datos que indiquen un beneficio de supervivencia y calidad de vida vinculado a la detección e intervención tempranas.[34,35]
Debido a que estas lesiones tienden a crecer en forma lenta, muy pocas veces son mortales y su manifestación clínica tal vez se produzca muchos años después de la exposición a la radiación, hay preocupaciones importantes en cuanto a los costos y perjuicios de la sobredetección.[36]
Los grupos de expertos se abstienen de promover o disuadir el uso de la ecografía como instrumento de detección del cáncer de tiroides y esto continúa siendo un área de investigación activa.[37]
Después de una evaluación sistemática de la evidencia científica, el International Guideline Harmonization Group concluyó que el inicio de la vigilancia y la modalidad de vigilancia (palpación de la tiroides vs. ecografía) se deben determinar mediante una toma de decisiones compartida entre el proveedor de atención de la salud y el sobreviviente, después de una consideración minuciosa de los beneficios y los perjuicios. Además de sus recomendaciones, proporcionan ayuda para la toma de decisiones con el fin de facilitar las conversaciones sobre este tema.[34]
Para obtener información sobre los cánceres de tiroides subsiguientes, consultar la sección Neoplasias subsiguientes.
Disfunción tiroidea posterior a un trasplante
Los sobrevivientes de trasplante de células madre hematopoyéticas (TCMH) en la niñez tienen un aumento del riesgo de disfunción tiroidea.[38,39]
En un estudio retrospectivo de una sola unidad de trasplante pediátrico, se observó a 244 pacientes pediátricos durante un período de 20 años. Estos pacientes se habían sometido a TCMH por enfermedades malignas o no malignas entre 1999 y 2018. Los pacientes se evaluaron con un mínimo de 4 pruebas de función tiroidea y al menos 1 ecografía tiroidea de forma secuencial después del TCMH.[39]
Se detectaron complicaciones tiroideas en el 19,7 % de los pacientes.
La incidencia acumulada a 15 años de disfunciones tiroideas autoinmunitarias o no autoinmunitarias (34 %) no difirió estadísticamente entre los pacientes que recibieron regímenes a base de irradiación corporal total (ICT) y los pacientes que recibieron regímenes a base de quimioterapia (P = 0,23). La incidencia acumulada después del acondicionamiento a base de busulfano fue similar a la del ICT (incidencia acumulada a 10 años, 22,2 vs. 25,9 %, respectivamente).
La incidencia acumulada de hipotiroidismo no autoinmunitario fue estadísticamente más alta después del acondicionamiento a base de busulfano (12,4 %), que después de otros regímenes de acondicionamiento a base de quimioterapia sola (3,1 %; P = 0,02, incidencia acumulada a 5 años).
La incidencia general de nódulos tiroideos fue baja durante los primeros 5 años después del TCMH (1,9 %), pero aumentó de manera pronunciada con el tiempo, con una incidencia acumulada a 15 años de hasta el 52,1 %. Los pacientes que recibieron regímenes de acondicionamiento a base de ICT tuvieron una incidencia acumulada de nódulos a 15 años más alta (66,8 %) que los pacientes que recibieron regímenes de quimioterapia sola (33,6 %; P = 0,02).
La edad mayor de 10 años en el momento del trasplante mostró un efecto protector (CRI, 0,42).
El seguimiento ecográfico mostró una reducción progresiva estadísticamente significativa del diámetro anteroposterior tiroideo en los pacientes que recibieron regímenes de acondicionamiento basados en ICT (P = 0,005), pero no en los que recibieron regímenes de quimioterapia sola.
En un informe del Fred Hutchinson Cancer Research Center, el riesgo de disfunción tiroidea después de un TCMH fue mucho más bajo (15–16 %) después de irradiación corporal total (ICT) fraccionada, en comparación con una sola dosis de ICT (46–48 %).[38]
El aumento del riesgo de disfunción tiroidea no difirió entre los niños que recibieron un régimen con ICT o un régimen a base de busulfano (P = 0,48).
No se han estudiado otras terapias de dosis altas.
La deficiencia de TSH/TRH (hipotiroidismo de origen central) se describe con los efectos tardíos que afectan la hipófisis.
En el Cuadro 9 se resumen los efectos tardíos tiroideos y los exámenes médicos de detección correspondientes.
Cuadro 9. Efectos tardíos tiroideosa
Tratamiento predisponente
Efectos endocrinos o metabólicos
Exámenes médicos de detección
131I-MIBG = yodo I 131-metaiodobencilguanidina; T4 = tiroxina; TSH = hormona estimulante de la tiroides.
Radioterapia con exposición de la glándula tiroidea; tiroidectomía
Hipotiroidismo primario
Concentración de TSH
Radioterapia con exposición de la glándula tiroidea
Hipertiroidismo
Concentración de T4 libre
Concentración de TSH
Radioterapia con exposición de la glándula tiroidea, incluye 131I-MIBG
Nódulos tiroideos
Examen de la tiroides
Ecografía tiroidea
Sistema hipotalamohipofisario
Los sobrevivientes de cáncer infantil están en riesgo de padecer una serie de anomalías neuroendocrinas, principalmente debido al efecto de la radioterapia dirigida al hipotálamo.
Hay 2 categorías de sobrevivientes de cáncer infantil que experimentan disfunción hipotálamo-hipofisaria después de la radiación: los sobrevivientes con un tumor del SNC y los sobrevivientes tratados por un tumor de cabeza o cuello que no afectan el SNC o leucemia, o las personas con antecedentes de TCMH.[40]
Además, es posible que la presentación de tumores o la resección quirúrgica cerca del hipotálamo o de la hipófisis produzcan daño anatómico directo a estas estructuras, y disfunción hipotalámica o hipofisaria.
En esencia, todo el sistema hipotalamohipofisario está en riesgo.[41-43]
La calidad de la bibliografía relativa a la endocrinopatía hipofisaria en los sobrevivientes de cáncer infantil suele estar limitada por la recopilación retrospectiva de datos, el tamaño pequeño de la muestra, el sesgo de selección y participación de la cohorte, la heterogeneidad en el abordaje del tratamiento, el tiempo transcurrido desde el tratamiento y el método de comprobación. No obstante, la evidencia que vincula este desenlace con la radioterapia, la cirugía y la infiltración tumoral es convincente porque las personas afectadas, por lo general, presentan anomalías metabólicas y de desarrollo en las primeras etapas del seguimiento.
El riesgo de disfunción hipotálamo-hipofisaria aumenta con dosis más altas de radioterapia. Cuando la dosis de radioterapia supera los 30 Gy, hay un riesgo más alto de presentar trastornos hipofisarios hipotálamos, como deficiencia de la hormona adrenocorticotrópica (ACTH), deficiencia de la hormona luteinizante (LH) o de la hormona foliculoestimulante (FSH) y deficiencia de TSH.[40]
Diabetes insípida de origen central
Es posible que la diabetes insípida de origen central anuncie el diagnóstico de craneofaringioma, tumor de células germinativas supraselar o histiocitosis de células de Langerhans.[44-46]
La diabetes insípida a veces se manifiesta como una deficiencia aislada de la hipófisis en el momento de la presentación de tumores selares o supraselares, si bien con la progresión del tumor quizás se presenten otras deficiencias de hormonas hipofisarias.
La diabetes insípida se presenta con más frecuencia en el marco de un panhipopituarismo, que obedece a los efectos del tumor en el sistema hipotalamohipofisario-adrenal (HHA) o es consecuencia de procedimientos quirúrgicos realizados para lograr el control local del tumor.
Es poco probable que la diabetes insípida central se presente como un efecto tardío de la resección quirúrgica del tumor después de 2 años de seguimiento.[47] Su aparición después de ese período de tiempo debe motivar la evaluación de cambios tumorales u otras causas.
No se ha notificado diabetes insípida de origen central como efecto tardío de la irradiación craneal administrada a sobrevivientes de cáncer infantil.[3]
Deficiencia hormonal de la adenohipófisis
Las deficiencias hormonales de la adenohipófisis y de los factores reguladores hipotalámicos más importantes son efectos tardíos comunes en los sobrevivientes tratados con irradiación craneal.[43]
Evidencia (prevalencia de deficiencia hormonal de la adenohipófisis):
Los pacientes jóvenes con tumores encefálicos son particularmente vulnerables a la disfunción hipotalámico-hipofisaria. En un estudio participaron 718 sobrevivientes de tumores de encéfalo infantiles que se controlaron durante una mediana de 7,9 años.[48]
No se observaron diferencias en la prevalencia de la disfunción hipotalámico-hipofisaria en lactantes (0–1 años), niños pequeños (≥1–3 años) o grupos de edad mayores en el momento del diagnóstico, a pesar del hecho de que la radioterapia se usó con menos frecuencia en lactantes.
La exposición a la radiación (OR, 16,44; IC 95 %, 8,93–30,27), la ubicación del tumor supraselar (OR, 44,76; IC 95 %, 19,00–105,49) y la edad más temprana (OR, 1,11; IC 95 %, 1,05–1,18) se relacionaron con disfunción hipotalámica.
Durante el seguimiento, los lactantes y los niños pequeños sobrevivientes de tumores de encéfalo mostraron más aumento de peso (P < 0,0001) y una puntuación de desviación estándar (PDE) de estatura más pequeña (P = 0,001) que los sobrevivientes diagnosticados a una edad más avanzada.
Entre los sobrevivientes no irradiados, los lactantes y niños pequeños en el momento del diagnóstico recibieron un diagnóstico significativamente más frecuente de deficiencia de TSH, ACTH y hormona antidiurética (HAD), en comparación con los sobrevivientes diagnosticados a una edad más avanzada, probablemente causada por terapias sistémicas o el trauma de la neurocirugía.
En un estudio dirigido a datos más refinados sobre las dosis de radiación relacionadas con la insuficiencia hipofisaria, se analizaron 102 pacientes tratados por tumores que incluyeron exposiciones a radioterapia (SNC, cabeza y cuello, neoplasias malignas hematológicas) entre 2008 y 2019. La mediana de edad (intervalo intercuartílico [IIC]) en el momento de la radioterapia fue de 9,0 años (6,0–12,0 años) y la mediana de seguimiento fue de 5,7 años (3,5–9,1 años). La mayoría de los pacientes recibieron radioterapia focal encefálica (36,3 %) o ICT (32,4 %). Los diagnósticos más frecuentes fueron LLA (25,5 %) y meduloblastoma (17,6 %).[49]
La mayoría de los pacientes presentaron insuficiencia hipofisaria (64 pacientes; 62,7 %). De los pacientes, el 41 % presentaba una deficiencia hormonal y el 38 % tenía 2 deficiencias hormonales.
La deficiencia de la hormona del crecimiento (58 pacientes; 56,9 %) y la deficiencia de TSH (32 pacientes; 31,4 %) fueron las más comunes.
Los pacientes que presentaron insuficiencia hipofisaria recibieron una dosis máxima hipofisaria más alta (mediana [IIC], 44,0 Gy [20,4–54,0] vs. 18,2 Gy [14,4–52,6]; P = 0,008). Las dosis de 40 Gy a 49 Gy o más de 50 Gy produjeron un cociente de incidencia acumulada más alto que las dosis inferiores a 20 Gy (CRI, 4,07; P < 0,001 y CRI, 3,04; P < 0,001, respectivamente).
Sin embargo, incluso en bandas de dosis más bajas, los niveles de insuficiencia hipofisaria fueron significativos. La incidencia acumulada a 5 años fue superior al 30 % para la deficiencia de la hormona del crecimiento con dosis inferiores a 20 Gy y para la deficiencia de TSH con dosis de 20 a 29 Gy.
En un estudio de una sola institución, se controló a 1713 adultos sobrevivientes de cánceres y tumores de encéfalo infantiles (mediana de edad, 32 años) para una mediana de seguimiento de 25 años.[42]
La prevalencia de los trastornos del sistema hipotalamohipofisario fue del 56,4 % en aquellos expuestos a radioterapia craneal con dosis de 18 Gy o más.
En un estudio se hizo una evaluación sistemática a 3141 sobrevivientes de cáncer infantil para detectar trastornos hipotalamohipofisarios durante una mediana de seguimiento de 24,1 años:[50]
En los pacientes que recibieron radioterapia hipotalamohipofisaria (n = 1089), se observó lo siguiente:
La prevalencia estimada de deficiencia de hormona del crecimiento fue del 40,2 %.
La prevalencia estimada de deficiencia de TSH fue del 11,1 %.
La prevalencia estimada de deficiencia de LH/FSH fue del 10,6 %.
La prevalencia estimada de deficiencia de ACTH fue del 3,2%.
La prevalencia estimada de pubertad precoz de origen central fue del 0,9 %.
En los pacientes que no recibieron radioterapia hipotalamohipofisaria, la prevalencia estimada para la deficiencia de hormona del crecimiento fue del 6,2 %, y la prevalencia fue inferior al 1 % para los otros trastornos hipotalamohipofisarios.
Los factores clínicos relacionados de forma independiente con trastornos hipotalamohipofisarios fueron los siguientes:
Radioterapia hipotalamohipofisaria (incluso con dosis relativamente bajas, a excepción de la deficiencia de ACTH que no se observó hasta >30 Gy).
Alquilantes (deficiencia de hormona del crecimiento, deficiencia de LH/FSH).
Quimioterapia intratecal (deficiencia de hormona del crecimiento).
Hidrocefalia con colocación de una derivación (deficiencia de hormona del crecimiento, deficiencia de LH/FSH).
Convulsiones (deficiencia de TSH, deficiencia de ACTH).
Accidente cerebrovascular (deficiencia de hormona del crecimiento, deficiencia de TSH, deficiencia de LH/FSH, deficiencia de ACTH).
Los desenlaces adversos relacionados con trastornos hipotalamohipofisarios fueron los siguientes:
Estatura baja (deficiencia de hormona del crecimiento, deficiencia de TSH).
Carencia mineral ósea grave (deficiencia de hormona del crecimiento, deficiencia de LH/FSH).
Obesidad (deficiencia de LH/FSH).
Debilidad (deficiencia de hormona del crecimiento).
Deterioro de la calidad de vida relacionada con la salud física (deficiencia de TSH).
Disfunción sexual (deficiencia de LH/FSH).
Deterioro de la memoria y la velocidad de procesamiento (deficiencia de hormona del crecimiento, deficiencia de TSH).
El International Late Effects of Childhood Cancer Guideline Armonization Group (IGHG), formado por 42 expertos internacionales de varias disciplinas, realizó una búsqueda sistemática de bibliografía sobre la vigilancia del sistema hipotalamohipofisario en sobrevivientes de cáncer infantil.[40]
Se observó que la exposición a la radiación del sistema hipotalamohipofisario aumentó el riesgo de disfunción hipotalámico-hipofisaria, con una relación dependiente de la dosis. Las dosis de radioterapia superiores a 18 Gy se relacionan con un riesgo más alto de deficiencia de la hormona del crecimiento y pubertad precoz central.
Por lo general, las deficiencias en ACTH, TSH y LH/FSH se presentan después de administrarse dosis superiores a 30 Gy.
En el panel se observó que los sobrevivientes de cáncer infantil con lesión tisular local en la región hipotalámico-hipofisaria corren el riesgo de presentar disfunción hipotalámico-hipofisaria desde el momento del diagnóstico o la cirugía por un tumor encefálico ubicado cerca o dentro de la región hipotalámico-hipofisaria, o por la aparición de hidrocefalia (en particular, con riesgo de deficiencia de la hormona del crecimiento y pubertad precoz de origen central).
Las personas expuestas a la radiación corren el riesgo de presentar disfunción hipotalámico-hipofisaria un año después de la finalización de la radioterapia.
Los sobrevivientes de cáncer infantil siguen en riesgo de deficiencia de la hormona del crecimiento durante al menos 15 años después del diagnóstico de cáncer o la exposición al tratamiento. El riesgo más allá de 15 años depende de los factores individuales del paciente. Para la pubertad precoz de origen central, los pacientes permanecen en riesgo hasta los 8 años en las niñas y los 9 años en los niños.
Las 6 hormonas de la adenohipófisis y sus principales factores reguladores hipotalámicos se presentan en el Cuadro 10.
Cuadro 10. Hormonas de la adenohipófisis y principales factores reguladores hipotalámicos
Hormona hipofisaria
Factor hipotalámico
Regulación hipotalámica de la hormona hipofisaria
(–) = inhibidora; (+) = estimuladora.
Hormona del crecimiento (GH)
Hormona liberadora de la hormona de crecimiento
+
Somatostatina
–
Prolactina
Dopamina
–
Hormona luteinizante (LH)
Hormona liberadora de gonadotropina
+
Hormona foliculoestimulante (FSH)
Hormona liberadora de gonadotropina
+
Hormona estimulante de la tiroides (TSH)
Hormona liberadora de la hormona estimulante de la tiroides
+
Somatostatina
–
Hormona adrenocorticotropa (ACTH)
Hormona liberadora de corticotropina
+
Vasopresina
+
Deficiencia de la hormona del crecimiento
La deficiencia de la hormona del crecimiento es la más común y, a menudo, la primera deficiencia de la adenohipófisis que se presenta después de la radioterapia craneal. En los sobrevivientes infantiles de tumores del SNC, la prevalencia general de deficiencia de la hormona del crecimiento es del 12,5 %.[3]
El riesgo aumenta con la dosis de radiación y el tiempo transcurrido desde el tratamiento.
La deficiencia de la hormona del crecimiento quizás sea ocasionada por dosis relativamente bajas de radiación craneal.[51]
Es posible que la deficiencia de la hormona del crecimiento se presente después de la administración de 18 a 24 Gy de radiación craneal, que se usa para tratar a pacientes con LLA y linfoma. Cuanto más alta sea la dosis de radiación, antes se producirá una deficiencia de la hormona del crecimiento después del tratamiento.[52]
Entre el 60 % y el 80 % de los pacientes pediátricos con tumores de encéfalo que recibieron dosis de radioterapia superiores a 30 Gy tendrá una disminución de la respuesta de la hormona del crecimiento sérica frente a una prueba de estimulación, por lo general dentro de los 5 años a partir del tratamiento.[53,54]
Evidencia (relación entre la dosis de radiación y la respuesta de la deficiencia de la hormona del crecimiento en sobrevivientes de tumores de encéfalo infantiles):
En un informe de 156 pacientes con meduloblastoma que se trataron entre 2003 y 2013 en el estudio SJMB03, se observó lo siguiente:[55]
La incidencia acumulada a 5 años de la deficiencia de la hormona de crecimiento fue del 68,9 %, y la incidencia acumulada del reemplazo de la hormona de crecimiento fue del 48,1 %.
La deficiencia de la hormona de crecimiento se relacionó con una mayor dosis en el hipotálamo (CRI, 1,035; P = 0,0055) en los pacientes de riesgo medio y de riesgo alto que tenían una derivación del líquido cefalorraquídeo (CRI, 2,532; P = 0,0049).
En un estudio de radioterapia conformada (RTC) en niños con tumores del SNC, se indica que la deficiencia de la hormona del crecimiento por lo general se presenta en los 12 meses posteriores a la radioterapia, según los efectos de la dosis y el volumen en el hipotálamo.[56]
En un informe con datos de 118 pacientes de tumores de encéfalo localizados tratados con radioterapia, se modeló la concentración máxima de hormona del crecimiento como una función exponencial del tiempo posterior a la RTC y la dosis media de radiación dirigida al hipotálamo.[54]
Se pronosticó que el paciente promedio presentará deficiencia de la hormona del crecimiento con las siguientes combinaciones del tiempo posterior a la RTC y la dosis media dirigida al hipotálamo: 12 meses y más de 60 Gy; 36 meses y 25 Gy a 30 Gy, y 60 meses y 15 Gy a 20 Gy.
Una dosis acumulada de 16,1 Gy dirigida al hipotálamo se consideraría la dosis media de radiación necesaria para alcanzar un riesgo del 50 % de deficiencia de hormona del crecimiento a 5 años (DT50/5) (consultar la Figura 7).
En una revisión del PENTEC se informó acerca del riesgo de deficiencia de la hormona de crecimiento en sobrevivientes de cáncer infantil que se trataron con radioterapia.[20]
Mediante el modelo de probabilidad de complicaciones del tejido normal para la deficiencia de la hormona de crecimiento (18 cohortes, 545 pacientes) se encontró que 24,9 Gy era el D50 (IC 95 %, 20,9–28,0).
Mediante el modelo de probabilidad de complicaciones del tejido normal ajustado para la irradiación encefálica total en niños con una mediana de edad superior a 5 años se indicó un 20 % de riesgo de deficiencia de la hormona de crecimiento en los pacientes que recibieron una dosis media de 21 Gy en fracciones de 2 Gy en el eje hipotalamohipofisario.
Evidencia (riesgo de deficiencia del crecimiento en sobrevivientes de cáncer infantil):
En un estudio se evaluó a 127 pacientes de LLA que recibieron dosis de radioterapia craneal de 24 Gy, 18 Gy o que no recibieron este tipo de radioterapia (época del tratamiento, 1980–1990).[57]
El cambio en la estatura, en comparación con las normas poblacionales expresadas como PDE, fue significativo para los 3 grupos. La respuesta a la dosis fue de -0,49 ± 0,14 para el grupo que no recibió radioterapia, -0,65 ± 0,15 para el grupo que recibió 18 Gy de radioterapia y -1,38 ± 0,16 para el grupo que recibió 24 Gy de radioterapia.
Los sobrevivientes de LLA infantil tratados con quimioterapia sola también tienen un aumento de riesgo de estatura baja en la edad adulta, aunque el riesgo más alto es el de aquellos tratados con radiación craneal o craneoespinal a una edad temprana.[58] En un estudio transversal, se determinó la estatura adulta alcanzada por 2434 sobrevivientes de LLA que participaron en el CCSS.
Todos los grupos de sobrevivientes expuestos a tratamiento (quimioterapia sola, y quimioterapia con radioterapia craneal o craneoespinal) tuvieron una disminución de la estatura adulta y un riesgo mayor de estatura baja en la edad adulta (PDE de la estatura < -2), en comparación con los hermanos (P < 0,001).
El riesgo de estatura baja para los sobrevivientes tratados con quimioterapia sola fue alto en comparación con los hermanos (OR, 3,4; IC 95 %, 1,9–6,0).
Entre los sobrevivientes, los factores de riesgo significativos de estatura baja fueron el diagnóstico de LLA antes de la pubertad, las dosis más altas de radiación craneal (≥20 vs. <20 Gy), cualquier tipo de radioterapia dirigida a la columna vertebral (porque afecta los cuerpos vertebrales) y el sexo femenino.
El efecto de la quimioterapia sola en el crecimiento de 67 sobrevivientes tratados con regímenes contemporáneos para la LLA fue estadísticamente significativo a -0,59 de DE. La pérdida de potencial de crecimiento no se correlacionó con el estado de la hormona del crecimiento en este estudio, lo que destaca aún más la participación de otros factores en las deficiencias de crecimiento observadas en esta población.[59]
En un estudio longitudinal con 372 sobrevivientes de LLA tratados en una sola institución en el contexto de un ensayo de solo quimioterapia, se observaron los siguientes resultados:[60]
Las puntuaciones z de estatura disminuyeron durante el tratamiento y mejoraron después de la terapia.
Una edad más baja en el momento del diagnóstico (2 a <10 años), el estado de LLA de riesgo bajo, el recuento de glóbulos blancos menor de 50 × 109/l en el momento del diagnóstico, o el estado negativo para el SNC se relacionaron con mejorías significativas en las puntuaciones z para la estatura durante el período sin tratamiento en comparación con los pacientes de más edad en el momento del diagnóstico (edad ≥10 años), que tenían estado de LLA de riesgo estándar o de riesgo alto, un recuento de glóbulos blancos de 50 × 109/l o más alto, o un estado positivo para el SNC.
La pérdida de estatura potencial en los pacientes de más edad se atribuyó a la atenuación del período de crecimiento rápido durante el tratamiento, sin recuperación después de este, y a la intensidad de la quimioterapia en pacientes con características de riesgo estándar o riesgo alto.
En un estudio francés del CCSS se evaluaron los factores de riesgo de estatura baja en el adulto y la deficiencia de la hormona del crecimiento en 2965 sobrevivientes de cáncer infantil utilizando los datos clínicos y de tratamiento disponibles en las historias clínicas. Se excluyeron los pacientes que recibieron hormonas del crecimiento (6,4 %).[61]
Los factores de riesgo independiente para la estatura baja en la edad adulta fueron: ser joven en el momento del tratamiento del cáncer (RR, 0,9 por cada año de edad), presentar una estatura baja en el momento del diagnóstico (≤2 PDE: RR, 6,7), y haber recibido radioterapia dirigida a la hipófisis (5–20 Gy: RR, 4,2; 20–40 Gy: RR, 10,2; ≥ 40 Gy: RR, 19,5), busulfano (RR, 4,5) o más de 300 mg/m2 de lomustina (300–600 mg/m2: RR, 4,2 y ≥600 mg/m2: RR, 9,1).
Los niños que se trataron con una radiación de 15 Gy o más dirigidas a un volumen de 90 % o mayor de 7 vértebras y que no recibieron irradiación en la hipófisis tuvieron un aumento del riesgo de estatura baja (RR, 4,6). Este riesgo aumentó 1,3 a 2,4 veces si además recibieron radiación dirigida a la hipófisis.
En una investigación del St. Jude Children's Research Hospital se evaluaron los desenlaces de estatura a largo plazo (mediana de seguimiento, 10,2 años) entre 212 pacientes pediátricos con tumores embrionarios del SNC que se trataron con ICE de 23,4 Gy (n = 147), 36 Gy o más (n = 65).[62]
En comparación con los estándares de los Estados Unidos, la media de las puntuaciones z de estatura final a los 18 años fue de -1,3 para las mujeres sobrevivientes y de -1,5 para los varones sobrevivientes.
Los factores relacionados con el riesgo de deficiencia de estatura fueron la edad más joven en el momento de la ICE, una dosis más alta de ICE y el sexo femenino.
Los factores relacionados con tasas de crecimiento más altas antes de los 15 años fueron la edad avanzada en el momento de la ICE, el sexo masculino, las dosis de ICE menores a 36 Gy, la terapia de reemplazo para la hormona del crecimiento y para la insuficiencia suprarrenal central, y la raza blanca.
Crecimiento después de un trasplante de células madre hematopoyéticas
Los niños que se someten a un trasplante de células madre hematopoyéticas (TCMH) con ICT tienen un riesgo significativo de deficiencia de la hormona del crecimiento y de los efectos directos de la radiación en el desarrollo esquelético.[63-65]
El riesgo de deficiencia de la hormona del crecimiento aumenta con dosis de ICT únicas en lugar de fraccionadas, irradiación craneal administrada antes del trasplante, sexo femenino y complicaciones posteriores al tratamiento, como enfermedad de injerto contra huésped (EICH).[63-65]
El hiperfraccionamiento de la dosis de ICT reduce en gran medida el riesgo para pacientes que no se sometieron a irradiación craneal para la profilaxis o el tratamiento de la leucemia en el SNC.[66]
En algunos estudios, los regímenes que contienen busulfano y ciclofosfamida aumentaron el riesgo,[65,67] pero no en otros.[68]
Evidencia (deficiencia de la hormona del crecimiento en sobrevivientes de TCMH en la niñez):
En el Late Effect Working Party del European Group for Blood and Marrow Transplantation, se estudiaron y analizaron los efectos tardíos que se presentan después de un TCMH. Los siguientes resultados se observaron en 181 pacientes de anemia aplásica, leucemias y linfomas sometidos a un TCMH antes de la pubertad:[69,70]
Disminución general en el valor de PDE de la estatura final en comparación con la estatura en el momento del trasplante y la estatura genética. Se calcula que la media de pérdida de estatura es de casi 1 PDE de la estatura (6 cm) en comparación con la media de estatura en el momento del TCMH y la media de la estatura genética.
El tipo de trasplante, la EICH y el tratamiento con hormona del crecimiento o corticoesteroides no influyeron en la estatura final.
Se encontró que la ICT (radioterapia de dosis única más que la radioterapia de dosis fraccionada), el sexo masculino y la edad temprana en el momento del trasplante fueron factores importantes para la pérdida de estatura a largo plazo.
La mayoría de los pacientes (140 de 181) alcanzaron una estatura adulta en el intervalo normal de la población general.
Se notificó deficiencia de la hormona del crecimiento después de dosis más bajas de fracción única de 10 Gy y dosis fraccionadas de 12 a 18 Gy de ICT.[71]
Terapia de reemplazo de la hormona del crecimiento
La terapia de remplazo de la hormona del crecimiento proporciona el beneficio de lograr resultados de estatura óptimos en los niños que no han alcanzado la madurez esquelética.[43] El diagnóstico requiere pruebas dinámicas especializadas.[43,72]
El tratamiento con hormona del crecimiento recombinante (rGH) se suele postergar hasta 12 meses después de finalizar con éxito los tratamientos del cáncer o de un tumor de encéfalo, y después de un análisis multidisciplinario en el que participen el endocrinólogo pediatra a cargo de las recetas, el oncólogo principal y otros proveedores seleccionados por el paciente o la familia.[73]
Las preocupaciones concernientes a la inocuidad de la rGH en sobrevivientes de cáncer infantil se relacionaron principalmente con el potencial mitogénico de la hormona del crecimiento con estimulación del crecimiento tumoral en una población en riesgo alto de segundas neoplasias.[74] Sin embargo, la mayoría de los estudios que notifican estos desenlaces se ven limitados por el sesgo de selección y el tamaño pequeño de la muestra.
Evidencia (riesgo de neoplasia subsiguiente después de terapia de reemplazo de la hormona del crecimiento):
En un estudio, en el que se evaluó a 361 sobrevivientes de cáncer inscritos en el CCSS y tratados con la hormona del crecimiento, se comparó el riesgo de recidiva, el riesgo de una neoplasia subsiguiente y el riesgo de muerte entre los sobrevivientes que recibieron tratamiento con la hormona del crecimiento y los que no la recibieron.[75]
El RR de recidiva de la enfermedad fue de 0,83 (IC 95 %, 0,37–1,86) para los sobrevivientes tratados con la hormona del crecimiento.
Los participantes tratados con la hormona del crecimiento recibieron diagnósticos de 15 neoplasias malignas subsiguientes, todos tumores sólidos, con un RR general de 3,21 (IC 95 %, 1,88–5,46), sobre todo por un excedente pequeño de neoplasias subsiguientes observadas en los sobrevivientes de leucemia aguda.[75]
Con un seguimiento prolongado, disminuyó la elevación del riesgo de un cáncer subsiguiente resultante de la hormona del crecimiento.[76]
En comparación con los sobrevivientes que no recibieron hormona del crecimiento, aquellos que la recibieron tuvieron un exceso doble de riesgo de presentar una neoplasia subsiguiente (RR, 2,15; IC 95 %, 1,33–3,47; P < 0,002); los meningiomas fueron las neoplasias que se observaron con mayor frecuencia (9 de 20 tumores).[75]
En un estudio del CCSS se notificó el riesgo de neoplasias subsiguientes en el SNC después de un período de seguimiento más largo.[77]
La razón de tasas ajustada de meningioma y gliomas en sobrevivientes de tumores del SNC tratados con la hormona del crecimiento fue de 1,0 (IC 95 %, 0,6–1,8; P = 0,94) cuando se comparó con el de los sobrevivientes de tumores del SNC que no se trataron con hormona del crecimiento, lo cual indica que las diferencias son mínimas entre los 2 grupos para este riesgo en particular.
En una revisión de los datos disponibles, se indica que el tratamiento con la hormona del crecimiento no se relaciona con un aumento del riesgo de progresión o recidiva de un tumor en el SNC, ni de una leucemia nueva o recidivante.[78]
En general, los datos que dan cuenta de neoplasias malignas subsiguientes entre sobrevivientes de cáncer infantil tratados con hormona del crecimiento se deberán interpretar con reserva debido al número reducido de episodios.[43,56,73-75,79,80]
Como se describe en una declaración de consenso de la Growth Hormona Research Society, las áreas de incertidumbre incluyen la inocuidad de la hormona del crecimiento en niños con afecciones predisponentes al cáncer, su uso en niños con deficiencia de hormona del crecimiento que reciben terapia de mantenimiento y la razón de riesgo-beneficio general en adultos sobrevivientes.[81]
Trastornos de la hormona luteinizante y la hormona foliculoestimulante
La radioterapia craneal puede afectar de manera adversa el desarrollo puberal.
Las dosis superiores a 18 Gy a veces producen pubertad precoz de origen central, mientras que las dosis mayores de 30 a 40 Gy quizás produzcan deficiencia de la hormona luteinizante (LH) o la hormona foliculoestimulante (FSH).[50]
Pubertad precoz de origen central
Prevalencia y factores de riesgo
La pubertad precoz de origen central es una de las disfunciones hipotalamohipofisarias más comunes; la prevalencia varía entre los estudios debido a la composición de la cohorte del estudio y el método de comprobación.[3,82,83]
En el estudio SJMB03, 156 niños y adolescentes con meduloblastoma se trataron con cirugía, irradiación craneoespinal de fotones adaptada al riesgo y quimioterapia de dosis intensiva.[55]
La incidencia acumulada de pubertad precoz a 5 años fue del 2,0 %.
Los niños con tumores que crecen cerca del sistema hipotalamohipofisario (incluso los pacientes de neurofibromatosis de tipo 1) o los tratados con radiación craneal son más vulnerables.[3]
En un estudio de 178 sobrevivientes de tumores de encéfalo infantiles (mediana de seguimiento de 6,6, años), hubo pubertad precoz en el 12,2 % de los sobrevivientes con una incidencia acumulada a 5 años del 4,0 %.[3]
Se notificó pubertad precoz de origen central en algunos niños que recibieron irradiación craneal en dosis de 18 Gy o más.[82,84,85]
La hidrocefalia también aumenta el riesgo de pubertad precoz de origen central.[86]
Diagnóstico
La pubertad precoz de origen central se define por el comienzo del desarrollo puberal antes de los 8 años en las niñas y los 9 años en los varones como resultado de la activación prematura del sistema hipotalamohipofisario-gonadal.
No es posible realizar la evaluación de la pubertad con mediciones del volumen testicular en varones expuestos a quimioterapia o radiación directa a los testículos, dado el efecto tóxico de estos tratamientos en las células germinativas y las consecuencias en el tamaño gonadal.[87]
La estadificación de la pubertad en los varones de esta población se basa en la presencia de otros signos de virilización, como la aparición de vello púbico y la medición de las concentraciones plasmáticas de testosterona.[87]
Tratamiento y desenlaces relacionados con la pubertad precoz de origen central
Aparte del ajuste y los desafíos psicosociales relacionados con el desarrollo puberal precoz, la pubertad precoz a veces conduce al cierre rápido del cartílago de crecimiento esquelético y a una estatura baja. Este efecto perjudicial se puede potenciar aún más por una deficiencia de la hormona del crecimiento.[82,87]
El aumento de la velocidad de crecimiento inducido por el desarrollo puberal quizás oculta una deficiencia simultánea de la hormona del crecimiento con una velocidad de crecimiento en apariencia normal; en ocasiones, este hecho induce a error a los proveedores de atención.[87]
El efecto de la pubertad precoz de origen central en el crecimiento lineal se determina mediante la evaluación del grado de maduración esquelética (edad ósea) por medio de una radiografía de la mano izquierda.[88]
Para lograr retrasar el avance de la pubertad se usan diversas preparaciones de agonistas de la hormona liberadora de gonadotropina. Se ha demostrado que este abordaje mejora las perspectivas de crecimiento, en especial cuando se tratan al mismo tiempo otras anomalías hipofisarias como la deficiencia de la hormona del crecimiento.[43,89]
Deficiencia de la hormona luteinizante o la hormona foliculoestimulante
La deficiencia de la hormona luteinizante (LH) o la hormona folículo estimulante (FSH), también conocida como hipogonadismo hipogonadotrópico, quizás se manifieste con retraso puberal (después de los 13 años en las niñas y los 14 años en los niños), interrupción de la pubertad o síntomas de disminución de la producción de hormonas sexuales, según la edad y el estadio puberal en el momento del diagnóstico.
El riesgo de deficiencia de LH o FSH es más alto en los pacientes tratados con radiación craneal en dosis mayores o iguales a 30 Gy; es posible que la deficiencia de LH o FSH después de la exposición a dosis bajas ocurra en momentos posteriores. La deficiencia de LH o FSH después de la exposición a dosis más bajas se puede presentar en momentos de retraso.[4]
Con la administración de dosis más altas de radioterapia craneal (>35 Gy) se observan deficiencias de LH o FSH, con una incidencia acumulada del 10 % al 20 %, entre 5 y 10 años después del tratamiento.[4,86]
El tratamiento de estas deficiencias se hace con reemplazo de hormonas sexuales ajustado por edad y estado puberal.[43]
Deficiencia de la hormona estimulante de la tiroides o de la hormona liberadora de tirotropina
La deficiencia de la hormona estimulante de la tiroides (TSH) (también llamada hipotiroidismo de origen central) en los sobrevivientes de cáncer infantil tal vez tenga profundas consecuencias clínicas y esta subvalorada. Los sobrevivientes de tumores de cabeza y cuello que recibieron radioterapia con posible exposición de la región hipotalamohipofisaria y de la glándula tiroides, quizás presenten hipotiroidismo como resultado de la deficiencia de la hormona liberadora de tirotropina (TRH) o de la TSH (hipotiroidismo de origen central), disfunción de la glándula tiroides (hipotiroidismo primario) o una combinación de origen central y primario.
Prevalencia y factores de riesgo
El riesgo de deficiencia de TSH es más alto en los pacientes tratados con radiación craneal en dosis de 30 Gy o más. La deficiencia de TSH después de la exposición a dosis más bajas se puede presentar en momentos de retraso.[4]
La incidencia acumulada de la deficiencia de TSH fue del 23 % (±8 %) entre sobrevivientes de tumores de encéfalo embrionarios tratados con ICE adaptada al riesgo, irradiación conformada al sitio primario y quimioterapia de dosis alta. La dosis de radiación dirigida al hipotálamo superior a 42 Gy se relaciona con un aumento del riesgo de presentar deficiencia de TSH (44 ± 19 % para una dosis de ≥42 Gy y 11 % ± 8 % para una dosis de <42 Gy).[90]
En un informe de 156 niños y adolescentes con meduloblastoma que recibieron tratamiento entre 2003 y 2013 con cirugía, irradiación craneoespinal con fotones adaptada al riesgo y quimioterapia de dosis intensiva, la incidencia de hipotiroidismo fue del 48,4 %.[55]
En los pacientes de riesgo medio, el hipotiroidismo se relacionó con una edad más joven (CRI, 0,902; P = 0,0070), dosis en el hipotálamo (CRI, 1,039; P = 0,0273) y dosis en la tiroides (CRI, 1,070; P = 0,0263). La dosis media de radioterapia fue de 34,8 Gy (desviación estándar [DE], 7,5) en el hipotálamo y de 17,4 Gy (DE, 5,4) en la tiroides.
En los pacientes de riesgo alto, el hipotiroidismo se asoció con un aumento de la dosis en el hipotálamo (CRI, 1,068; P = 0,0671) y en la tiroides (CRI, 1,050; P = 0,0515). La dosis media de radioterapia fue de 47,8 Gy (DE, 4,87) en el hipotálamo y de 25,8 Gy (DE, 7,2) en la tiroides.
Los investigadores no pudieron distinguir de forma fiable entre hipotiroidismo de origen central y primario, pero estimaron que 8 de 85 pacientes fueron diagnosticados inicialmente con hipotiroidismo de origen central.
Entre los sobrevivientes de meduloblastomas tratados con radioterapia de fotones (mediana de seguimiento de 9,6 años) o protones (mediana de seguimiento de 3,8 años), se presentó hipotiroidismo primario en 12 de 54 pacientes (22 %) después de la radioterapia con fotones y en 3 de 41 pacientes (7 %) después de la radioterapia de protones (CRI, 2,1; P = 0,27). Se presentó hipotiroidismo de origen central (deficiencia de TSH) en 13 de 54 pacientes (24 %) después de la radioterapia con fotones y en 4 de 41 pacientes (10 %) después de la radioterapia de protones (CRI, 2,16; P = 18).[91]
De 118 sobrevivientes de meduloblastoma (mediana de seguimiento, 5,6 años), la incidencia acumulada estimada de hipotiroidismo fue del 35,8 % (IC 95 %, 26,7–47,1 %) 5 años después de la radioterapia.[18]
La mediana de tiempo desde el final de la radioterapia hasta que se presenta el hipotiroidismo fue de 2,1 años (intervalo, 0,1–7,1 años) después de la radioterapia de protones y 2,9 años (intervalo, 0,8–7,2 años) después de la radioterapia de fotones.
La tasa de hipotiroidismo (CRI, 2,36; IC 95 %, 1,11–5,02) fue mayor en los pacientes que se trataron con radioterapia de fotones que en aquellos que se trataron con radioterapia de protones.
Las diferencias en el riesgo de hipotiroidismo según la modalidad de radiación se atribuyeron sobre todo a tasas más elevadas de hipotiroidismo primario después de la radioterapia de fotones (CRI, 4,61; IC 95 %, 1,20–17,66).
Los pacientes con hipotiroidismo central se deben someter a pruebas de otras deficiencias hipotalamohipofisarias antes del reemplazo tiroideo. El reemplazo tiroideo puede aumentar la eliminación de cortisol, lo que agrava cualquier insuficiencia suprarrenal subyacente y contribuye a una posible crisis suprarrenal.
En un estudio de 189 niños y adultos jóvenes (edad <26 años) con tumores de encéfalo tratados con radioterapia de protones, la tasa actuarial a 4 años de hipotiroidismo fue del 20,1 %; el 90 % de la deficiencia de la TSH fue de origen central.[17]
Cuadro clínico inicial y diagnóstico
Los síntomas de hipotiroidismo de origen central (por ejemplo, astenia, edema, adormecimiento y sequedad de la piel) a veces son graduales y pasan desapercibidos hasta que se comienza una terapia de reemplazo tiroideo.
Además de demorar la pubertad y hacer más lento el crecimiento, el hipotiroidismo a veces causa fatiga, piel seca, estreñimiento, aumento de la necesidad de dormir e intolerancia al frío.
Las personas con deficiencia de TSH tienen concentraciones plasmáticas bajas de T4 libre y de TSH o concentraciones inadecuadamente normales de TSH. La disminución de las concentraciones de T4 dentro del intervalo normal también puede ser predictiva de la deficiencia de TSH. Los médicos no deben dudar en volver a evaluar a los sobrevivientes con este patrón de funcionamiento tiroideo, en especial en presencia de síntomas de hipotiroidismo.[92]
El hipotiroidismo primario mixto y de origen central también surge a raíz de lesiones separadas de la glándula tiroidea y el hipotálamo (por ejemplo, lesión por radiación a ambas estructuras), con lo cual se eleva un poco la concentración de la TSH a pesar de la contribución de las alteraciones hipotalamohipofisarias al hipotiroidismo en las personas afectadas.
Tratamiento de la deficiencia de la hormona estimulante de la tiroides
La terapia de reemplazo de la hormona tiroidea con levotiroxina es la base del tratamiento de la deficiencia de TSH.
La dosis de levotiroxina solo se debe ajustar según las concentraciones plasmáticas de T4 libre, ; y se anticipa que las concentraciones de TSH se mantengan bajas durante el tratamiento debido al origen central de este tipo de deficiencia.
El hipotiroidismo de origen central no se debe tratar antes de evaluar el funcionamiento de otras deficiencias hipotalámicas o hipofisarias porque es posible que precipite una insuficiencia suprarrenal aguda en pacientes con deficiencia de ACTH.
Deficiencia de ACTH
Prevalencia y factores de riesgo
La deficiencia de la hormona adrenocorticotrópica (ACTH) es menos frecuente que otras alteraciones neuroendocrinas, pero se deberá sospechar su presencia en pacientes con antecedentes de tumores de encéfalo (con independencia de la modalidad terapéutica), radioterapia craneal, deficiencia de la hormona del crecimiento o hipotiroidismo de origen central.[43,90,93,94]
La deficiencia de la ACTH se presenta casi con exclusividad en sobrevivientes que antes estuvieron expuestos a dosis hipotalámicas hipofisarias de 30 Gy o mayores.
La incidencia acumulada de la deficiencia de la ACTH fue del 38 % (±6 %) entre sobrevivientes de tumores de encéfalo embrionarios tratados con irradiación craneoespinal (ICE) adaptada al riesgo, irradiación conformada al sitio primario y quimioterapia de dosis alta. La dosis de radioterapia administrada al hipotálamo (mediana, 42,2 Gy para deficiencia no relacionada con la ACTH vs. 41,3 Gy para deficiencia de la ACTH) no afectó el estado de la ACTH.[90]
En un estudio de 189 niños y adultos jóvenes (edad <26 años) con tumores de encéfalo tratados con radioterapia de protones, la tasa actuarial a 5 años de deficiencia de la ACTH fue del 8 %, y se presentó casi en forma exclusiva después de dosis ≥40 Gy dirigidas al hipotálamo y la hipófisis.[17]
En un informe se incluyeron 156 niños y adolescentes con meduloblastoma que se trataron en el estudio SJMB03 con cirugía, irradiación craneoespinal de fotones adaptada al riesgo y quimioterapia de dosis intensiva. La incidencia de insuficiencia suprarrenal acumulada a 5 años fue del 13,0 %.[55] La insuficiencia suprarrenal se relacionó con una mayor dosis dirigida al hipotálamo (CRI, 1,112; P < 0,0001).
Si bien es poco frecuente, a veces se presenta una deficiencia de la ACTH en pacientes tratados con radiación intracraneal en dosis inferiores a 24 Gy y en pacientes con tumores que no afectan el SNC o neoplasias malignas hematológicas distantes de la región hipotalámico-hipofisaria. El uso de la terapia con glucocorticoides en los protocolos de tratamiento también se puede relacionar con insuficiencia suprarrenal inducida por esteroides, que en algunos casos puede ser prolongada.[95-97]
En un informe del PENTEC sobre deficiencia de la ACTH (6 cohortes, 230 pacientes), la dosis relacionada con un riesgo del 50 % fue de la de 61 Gy (IC 95 %, 44,7–119,4).[20] El riesgo de deficiencia de la ACTH fue del 20 % en los niños que recibieron una dosis media de 34 Gy en fracciones de 2 Gy dirigida al eje hipotalamohipofisario.
Diagnóstico y abordaje
Se deberá sospechar la deficiencia de la ACTH cuando las concentraciones plasmáticas de cortisol matutino son bajas (como examen de detección, una concentración de cortisol medida las 8 a.m. de 10 µg/dl o superior se considera compatible con la suficiencia de la ACTH, mientras que una concentración de 5 µg/dl o inferior hace sospechar la insuficiencia).
Se necesita confirmación mediante pruebas dinámicas, como la prueba de estimulación de la ACTH con dosis bajas.[93]
Debido al riesgo importante de insuficiencia suprarrenal de origen central en los sobrevivientes tratados con dosis de radiación craneal de 30 Gy dirigidos al sistema hipotalamohipofisario, se recomienda el seguimiento endocrino mediante pruebas dinámicas periódicas según lo indique la evaluación clínica para este grupo de riesgo alto.[43]
Los pacientes con una deficiencia parcial de la ACTH tal vez solo tengan síntomas leves a menos que padezcan de otra enfermedad.[43]
Es posible que una enfermedad altere la homeostasis habitual de estos pacientes y conlleve una evolución más grave, prolongada o complicada de lo esperado. Cualquier tipo de deficiencia de la ACTH, completa, incompleta o inadvertida, pone en peligro la vida del paciente cuando coincide con otra enfermedad.
El tratamiento de la deficiencia de la ACTH se basa en el reemplazo con hidrocortisona, que incluye dosis para situaciones de estrés por enfermedad con el fin de preparar el cuerpo a una mayor necesidad fisiológica de glucocorticoides.[43]
Hiperprolactinemia
Se describió la hiperprolactinemia en pacientes que recibieron dosis de radioterapia superiores a 50 Gy dirigidas al hipotálamo o que se sometieron a una cirugía que alteró la integridad del infundíbulo hipofisario.[98]
El hipotiroidismo primario quizás conduzca a hiperprolactinemia como resultado de una hiperplasia de tirotrofos y lactotrofos, presumiblemente debido a una hipersecreción de la TRH.
En estos pacientes, la respuesta de la prolactina a la TRH suele ser exagerada.[51,99]
La hiperprolactinemia en ocasiones genera retraso de la pubertad, galactorrea, irregularidades menstruales, pérdida de libido, crisis vasomotoras, esterilidad y osteopenia. La hiperprolactinemia como consecuencia de la radioterapia craneal casi nunca es asintomática, y con frecuencia se relaciona con el hipogonadismo (tanto de origen central como primario).
No es habitual que la hiperprolactinemia necesite tratamiento.[100]
En el Cuadro 11 se resumen los efectos tardíos hipofisarios y los exámenes médicos de detección correspondientes.
Cuadro 11. Efectos tardíos en la hipófisisa
Tratamiento predisponente
Efectos endocrinos o metabólicos
Exámenes médicos de detección
IMC = índice de masa corporal; FSH = hormona foliculoestimulante; LH = hormona luteinizante; T4 = tiroxina; TSH = hormona estimulante de la tiroides.
bLas mediciones del volumen testicular no son confiables para evaluar el desarrollo puberal en varones expuestos a quimioterapia o radiación directa a los testículos.
cSolo adecuada en el momento del diagnóstico. Las concentraciones de TSH no son útiles para el seguimiento durante la administración de terapia de reemplazo.
Tumor o cirugía que afecta el hipotálamo o la hipófisis. Radioterapia con exposición del sistema hipotalamohipofisario.
Deficiencia de la hormona del crecimiento
Evaluación del estado nutricional
Estatura, peso, IMC y estadio de Tannerb
Tumor o cirugía que afecta el hipotálamo, la hipófisis o las vías ópticas; hidrocefalia. Radioterapia con exposición del sistema hipotalamohipofisario.
Pubertad precoz
Estatura, peso, IMC y estadio de Tannerb
Concentraciones de FSH, LH, estradiol o testosterona
Tumor o cirugía que afecta el hipotálamo o la hipófisis. Radioterapia con exposición del sistema hipotalamohipofisario.
Deficiencia de gonadotropina
Antecedentes: pubertad y funcionamiento sexual
Hallazgos del examen: Estadio de Tannerb
Concentraciones de FSH, LH, estradiol o testosterona
Tumor o cirugía que afecta el hipotálamo o la hipófisis. Radioterapia con exposición del sistema hipotalamohipofisario.
Insuficiencia suprarrenal de origen central
Antecedentes: retraso del crecimiento, anorexia, deshidratación episódica, hipoglucemia, letargo e hipotensión sin causa aparente
Consulta endocrinológica para quienes recibieron una dosis de radiación ≥30 Gy
Radioterapia con exposición del sistema hipotalamohipofisario.
Hiperprolactinemia
Antecedentes o hallazgos del examen: galactorrea
Concentración de prolactina
Radioterapia con exposición del sistema hipotalamohipofisario.
Sobrepeso u obesidad
Estatura, peso e IMC
Evaluación de la presión arterial
Componentes de síndrome metabólico (obesidad abdominal, hipertensión, dislipidemia y deterioro del metabolismo de la glucosa)
Concentración de glucosa en ayunas y perfil de lípidos
Tumor o cirugía que afecta el hipotálamo o la hipófisis. Radioterapia con exposición del sistema hipotalamohipofisario.
Se ha observado un aumento de riesgo de síndrome metabólico o sus componentes en los sobrevivientes de cáncer infantil. La evidencia para este desenlace se ha obtenido por la notificación de afecciones clínicamente manifiestas en los sobrevivientes, por la evaluación retrospectiva de datos de historias clínicas y registros hospitalarios, así como de evaluaciones clínicas sistemáticas de cohortes bien caracterizadas desde el punto de vista clínico. Los estudios tienen limitaciones debido al sesgo de selección de la cohorte y de participación, la heterogeneidad en el enfoque de tratamiento, el tiempo desde el tratamiento y el método de confirmación. A pesar de estas limitaciones, existe evidencia concluyente que indica que el síndrome metabólico se relaciona estrechamente con episodios cardiovasculares y mortalidad.
Las definiciones del síndrome metabólico están en evolución, pero por lo general incluyen una combinación de obesidad de origen central (abdominal) con, por lo menos, 2 de las siguientes características:[101]
Hipertensión.
Dislipidemia aterógena (triglicéridos elevados y reducción del colesterol de lipoproteínas de densidad alta [HDL]).
Metabolismo anómalo de la glucosa (hiperglucemia en ayunas, hiperinsulinismo, resistencia a la insulina y diabetes mellitus de tipo 2).
Evidencia (prevalencia y factores de riesgo del síndrome metabólico en sobrevivientes de cáncer infantil):
Un grupo de investigadores evaluó la prevalencia y los determinantes del síndrome metabólico en la cohorte del estudio Dutch Childhood Cancer Survivors Study (DCCSS) LATER 2. Un total de 2338 sobrevivientes adultos de cáncer infantil se evaluaron y se compararon con 132 226 adultos de la cohorte de Lifelines. Estas personas no tenían antecedentes de cáncer.[102]
Los sobrevivientes tuvieron el doble de riesgo de presentar síndrome metabólico cuando se los comparó con los adultos sin antecedentes de cáncer. Este aumento del riesgo se produjo sobre todo por un aumento del riesgo de dislipidemia (aumento de triglicéridos o concentraciones bajas de HDL-C).
Se observó una prevalencia alta de síndrome metabólico (19,1 %) en esta cohorte de sobrevivientes jóvenes (mediana de edad, 34,7 años).
Otros factores relacionados con el síndrome metabólico fueron el consumo de tabaco (OR, 1,46), la baja actividad física (OR, 1,48), la radioterapia dirigida al abdomen (OR, 2,13; >30 Gy), la radioterapia dirigida al cráneo (OR para 1–25 Gy, 2,89; OR para >25 Gy, 2,44), la irradiación corporal total (OR, 6,17) y un tumor preexistente en el sistema nervioso central (OR, 1,78).
Un grupo de investigadores franceses evaluó la prevalencia general y específica por edad, así como los factores de riesgo del síndrome metabólico y sus componentes en 650 adultos sobrevivientes de leucemia infantil tratados sin TCMH.[103]
La prevalencia general de la afección fue del 6,9 %, según la siguiente prevalencia acumulada específica por edad:
20 años—1,3 %.
25 años—6,1 %.
30 años—10,8 %.
35 años—22,4 %.
La prevalencia de componentes individuales del síndrome metabólico fue la siguiente:
Aumento de glucemia en ayunas—5,8 %.
Aumento de triglicéridos—11,7 %.
Aumento de la circunferencia abdominal—16,7 %.
Disminución del colesterol HDL—26,8 %.
Aumento de la presión arterial—36,7 %.
Los factores clínicos que predijeron significativamente el riesgo de síndrome metabólico incluyeron el sexo masculino (OR, 2,64; IC 95 %, 1,32–5,29), la edad en la última evaluación (OR, 1,10, IC 95 %, 1,04–1,17) y el índice de masa corporal (IMC) en el momento del diagnóstico (OR, 1,15; IC 95 %, 1,01–1,32); pero no la dosis acumulada de esteroides. Los pacientes irradiados y los no irradiados exhibieron patrones distintos de anomalías metabólicas, con obesidad abdominal más frecuente en los pacientes irradiados e hipertensión más frecuente en los pacientes no irradiados.
En un estudio, se controló a 784 sobrevivientes a largo plazo de LLA infantil (mediana de edad, 31,7 años) durante una mediana de seguimiento de 26,1 años.[104]
La prevalencia de síndrome metabólico fue del 33,6 %, que fue significativamente mayor que en una cohorte de controles emparejados por edad, sexo y raza (n = 777) de la National Health and Nutrition Examination Survey (RR, 1,43; IC 95 %, 1,22–1,69).
Los factores de riesgo relacionados con el síndrome metabólico en este estudio incluyeron la edad avanzada y las exposiciones anteriores a radioterapia craneal.
Los componentes del síndrome metabólico, con una prevalencia significativamente mayor en los sobrevivientes de LLA que en los controles, incluyeron obesidad, resistencia a la insulina, hipertensión y disminución de las concentraciones de HDL.
En la cohorte francesa de sobrevivientes de leucemia infantil se observó que la prevalencia del síndrome metabólico aumentó entre los sobrevivientes de leucemia infantil (n = 1025), en comparación con los controles (n = 3202).[105]
Se encontró síndrome metabólico en 10,3 % de los pacientes (media de seguimiento, 16,3 años) y 4,5 % de los controles (OR, 2,49; P < 0,001). Los pacientes de trasplante que recibieron tratamiento con ICT presentaron el riesgo más alto (OR, 6,26; P < 0,001). Otros grupos de tratamiento que también mostraron un riesgo más alto que los controles fueron los pacientes tratados con quimioterapia sola. Las OR fueron de 1,68 (P = 0,005) después de la quimioterapia sola, 2,32 (P = 0,002) después de la quimioterapia y la irradiación craneal, y 2,18 (P = 0,057) después del trasplante sin irradiación.
En comparación con los controles, se observaron perfiles únicos en los sobrevivientes según el tratamiento. Los pacientes que recibieron ICT y presentaron síndrome metabólico tenían una circunferencia abdominal más pequeña que los controles. En contraste, los pacientes tratados con irradiación craneal y que presentaron síndrome metabólico tenían una circunferencia abdominal más grande que los controles. Además, los receptores de ICT presentaron concentraciones elevadas de triglicéridos, concentraciones de glucosa en ayunas y presión arterial sistólica.
En un estudio prospectivo de 164 sobrevivientes a largo plazo de tumores embrionarios tratados con radioterapia abdominal (mediana de seguimiento, 26 años), los sobrevivientes de nefroblastoma (OR, 5,2) y neuroblastoma (OR, 6,5) presentaron más componentes del síndrome metabólico que los controles.[106]
En comparación con los sobrevivientes que no recibieron radioterapia, los sobrevivientes tratados con irradiación abdominal tuvieron presión arterial, triglicéridos, colesterol de lipoproteínas de baja densidad (LDL) y porcentaje de grasa total más altos, según las mediciones por absorciometría dual de rayos X.
Repercusión del estilo de vida sobre los factores de riesgo modificables
Es posible que los factores de riesgo modificables (como la alimentación, la actividad física y los hábitos de consumo de tabaco) afecten el riesgo de presentar síndrome metabólico en los sobrevivientes de cáncer infantil.
En varios estudios se sustentaron los posibles beneficios de las modificaciones del estilo de vida para reducir el riesgo de enfermedad cardiovascular.[107-109]
Evidencia (modificaciones del estilo de vida para reducir el riesgo de enfermedad cardiovascular en sobrevivientes de cáncer infantil):
Los sobrevivientes que participaron en el estudio SJLIFE y que cumplieron con un estilo de vida saludable para el corazón presentaron un riesgo más bajo de síndrome metabólico.[107]
Las mujeres (RR, 2,4; IC 95 %, 1,7–3,3) y los varones (RR, 2,2; IC 95 %, 1,6–3,0) de la cohorte que no siguieron las recomendaciones alimentarias y las pautas de actividad física presentaron un exceso de riesgo de más del doble de presentar características clínicas de síndrome metabólico.
En una investigación del CCSS se evaluó el efecto del ejercicio en el riesgo de enfermedad cardiovascular entre sobrevivientes de linfoma de Hodgkin.[108]
El ejercicio vigoroso se relacionó con un riesgo más bajo de episodios cardiovasculares en forma dependiente de la dosis, sin importar el perfil de riesgo cardiovascular ni el tratamiento.
Los sobrevivientes que cumplieron con las directrices nacionales de ejercicio de intensidad vigorosa, presentaron una reducción del 51 % en el riesgo de presentar cualquier episodio cardiovascular, en comparación con aquellos que no siguieron las directrices.
En otra investigación del CCSS se evaluó la relación entre el ejercicio y la mortalidad en adultos sobrevivientes de cáncer infantil.[109]
Después de ajustar por las afecciones crónicas y las exposiciones de tratamiento, la mortalidad por todas las causas tuvo una relación inversa con los cuartiles del ejercicio notificado por los sobrevivientes (0, 3–6, 9–12, y 15–21 horas de equivalentes metabólicos de una tarea [MET]/semana).
Los sobrevivientes que cumplieron con los niveles recomendados de ejercicio vigoroso (≥9 horas de MET/semana) durante la adultez temprana y los que aumentaron el ejercicio durante 8 años, presentaron un riesgo de mortalidad más bajo.
Metabolismo anómalo de la glucosa
Para las personas tratadas a una edad temprana (edad <11 años), la radioterapia abdominal y la ICT se reconocen cada vez más como factores de riesgo independientes de diabetes mellitus en los sobrevivientes de cáncer infantil.[2,110-114]
Evidencia (factores de riesgo de diabetes mellitus en sobrevivientes de cáncer infantil):
En un estudio de cohortes de un solo centro con 532 adultos (mediana de seguimiento, 17,9 años) sobrevivientes a largo plazo (mediana de edad, 25,6 años), se observó lo siguiente:[111]
El tratamiento, pero no la variación genética, tuvo una relación estrecha con la presentación de componentes del síndrome metabólico.
Este síndrome fue más frecuente en los sobrevivientes que recibieron radiación craneal (23,3 %, P = 0,002) o abdominal (23,4 %, P = 0,009) que en los sobrevivientes que no la recibieron (10,0 %).
En un estudio transversal, se evaluaron los factores de riesgo cardiovascular y la resistencia a la insulina en una cohorte heterogénea, desde el punto de vista clínico, de 319 sobrevivientes de cáncer infantil durante 5 años o más desde el diagnóstico y 208 hermanos de control.[115]
La resistencia a la insulina fue significativamente más alta en los sobrevivientes tratados con cisplatino y radiación craneal (92 % de tumores de encéfalo) y en aquellos que recibieron corticoesteroides, pero no cisplatino (la mayoría de sobrevivientes de leucemia), en comparación con los hermanos.
La resistencia a la insulina no difirió entre los sobrevivientes tratados con cirugía sola y los hermanos.
Entre los sobrevivientes, el análisis de los fármacos quimioterapéuticos individuales no permitió encontrar relaciones con factores de riesgo cardiovascular o resistencia a la insulina.
Sin embargo, en comparación con sus hermanos, casi todos estos fármacos quimioterapéuticos (incluso cuando se analizan de manera individual) se relacionan con un perfil de riesgo cardiovascular alto, caracterizado por una masa corporal total magra más baja, un porcentaje alto de masa grasa y resistencia a la insulina.
En una cohorte europea multicéntrica de 2520 sobrevivientes de cáncer infantil (mediana de seguimiento, 28 años), se establecieron vínculos importantes entre la diabetes mellitus y el aumento de las dosis de radioterapia dirigidas a la cola del páncreas. Estos datos respaldan la contribución de la lesión de las células de los islotes inducida por la radiación a los deterioros de la homeostasis de la glucosa en esta población.[112]
En un informe del CCSS se comparó a 8599 sobrevivientes de cáncer infantil con 2936 hermanos del grupo de control seleccionados al azar, y se introdujeron ajustes por edad, el IMC y varios factores demográficos.[116]
El riesgo de diabetes mellitus fue 1,8 veces más alto en los sobrevivientes (IC 95 %, 1,03–2,05; P < 0,001).
Se encontraron relaciones significativas entre la diabetes mellitus y la edad temprana en el momento del diagnóstico (0–4 años), el uso de alquilantes, radioterapia abdominal o ICT.
Los sobrevivientes expuestos a radiación abdominal tuvieron un aumento de riesgo de diabetes 3,4 veces mayor (IC 95 %, 2,3–5,0; P < 0,001), en comparación con los hermanos y después de realizar el ajuste por IMC. En comparación con los hermanos, los sobrevivientes de neuroblastoma y tumor de Wilms tratados con radioterapia abdominal tuvieron un riesgo 6,9 y 2,1 veces mayor de diabetes, respectivamente. Las personas de estos grupos que no habían estado expuestas a la radioterapia abdominal no presentaron un riesgo mayor.
Los sobrevivientes tuvieron más probabilidad de recibir medicación por hipertensión, dislipidemia o diabetes mellitus que los hermanos del grupo de control.
En el Cuadro 12 se resumen los efectos tardíos de síndrome metabólico y los exámenes médicos de detección correspondientes.
Cuadro 12. Efectos tardíos del síndrome metabólicoa
Componentes de síndrome metabólico (obesidad abdominal, hipertensión, dislipidemia y deterioro del metabolismo de la glucosa)
Estatura, peso, IMC y evaluación de la presión arterial
Exámenes de laboratorio: glucosa y lípidos en ayunas
Composición corporal: peso insuficiente, sobrepeso y obesidad
Los sobrevivientes de cáncer infantil tienen riesgo de presentar una composición corporal anormal, que incluye peso insuficiente (IMC, <18,5), sobrepeso (IMC, >25,0 a IMC, <30,0) u obesidad (IMC, ≥30,0).
El IMC en el momento del diagnóstico se identificó como un factor pronóstico significativo de peso insuficiente o sobrepeso al realizar el seguimiento. Este hallazgo indica que los factores genéticos o ambientales contribuyen al desarrollo o la persistencia de una composición corporal anormal.[117,118]
Peso insuficiente
Alrededor de 1 de cada 10 sobrevivientes adultos de cáncer infantil tiene un peso inferior al normal. Los investigadores del CCSS descubrieron que los sobrevivientes de cáncer infantil con peso bajo tienen un mayor riesgo de mortalidad tardía.[119]
De los 9454 sobrevivientes (mediana de edad, 35 años; promedio de 17,5 años desde el diagnóstico), 627 (6,6 %) presentaron peso bajo en la encuesta inicial o en el cuestionario de seguimiento. De los 184 fallecidos, 29 tenían un peso inferior al normal al inicio del estudio.
El estado de delgadez fue más común entre las mujeres (9,1 vs. 4,5 %; P < 0,01), las personas con ingresos familiares más bajos (8,9 % para <$20 000 vs. 6 % para >$20 000; P < 0,01) y aquellas con antecedentes de una enfermedad crónica grave (P = 0,05). Tras ajustar por estos factores, junto con el consumo de tabaco previo y los antecedentes de radioterapia, los sobrevivientes con bajo peso presentaron un mayor riesgo de mortalidad por todas las causas en los 2 años siguientes a la notificación del IMC (OR, 2,85; P < 0,01), en comparación con los sobrevivientes con peso ideal.
Los investigadores del CCSS identificaron factores de riesgo relacionados con el tratamiento para el peso insuficiente; entre ellos la ICT (mujeres) o la irradiación abdominal (varones), el uso de alquilantes o antraciclinas.[118]
Sobrepeso u obesidad
Hasta la fecha, los pacientes de cáncer con un aumento de la incidencia de sobrepeso y obesidad son, sobre todo, sobrevivientes de LLA [117,120-124,41,98] y de tumores del SNC tratados con radioterapia craneal.[118,125]
La presentación de sobrepeso u obesidad después de recibir radioterapia craneal es multifactorial e incluye los siguientes aspectos:[122,126,127]
Deficiencia de la hormona del crecimiento.
Sensibilidad a la leptina.
Niveles reducidos de actividad física y gasto energético.
Evidencia (factores de riesgo de sobrepeso y obesidad):
En el estudio de cohorte nacional Dutch Childhood Cancer Survivor Study (DCCSS-LATER) de adultos sobrevivientes de cáncer infantil (n = 2338), los investigadores encontraron que casi la mitad de los sobrevivientes a largo plazo tenían exceso de peso (IMC >2,5 kg/m2). Esta incidencia no fue mayor que la de la población general.[128]
La incidencia del sobrepeso fue mayor en las mujeres de 50 años o más y la incidencia de obesidad mórbida fue superior en hombres de 50 años o más.
Factores como el exceso de peso en el momento del diagnóstico del cáncer, haber recibido irradiación craneal y tener deficiencia de la hormona de crecimiento se relacionaron con una condición de sobrepeso a largo plazo. El tratamiento con corticoesteroides no se relacionó con el sobrepeso.
Mediante la clasificación con absorciometría de rayos X de energía dual se identificó sobrepeso en un 30 % adicional de sobrevivientes, en particular aquellos que se trataron con irradiación dirigida al abdomen, ICT, antraciclinas y quimioterapia con derivados del platino.
Los investigadores del CCSS notificaron los siguientes factores de riesgo independientes para la obesidad en los sobrevivientes de cáncer infantil, entre ellos tratamiento, estilo de vida y consumo de medicamentos:[129]
Cáncer diagnosticado entre los 5 y 9 años de edad (RR, 1,12; IC 95 %, 1,01–1,24).
Funcionamiento físico anómalo (RR, 1,19; IC 95 %, 1,06–1,33).
Dosis de radiación hipotalámica o hipofisaria de 20 a 30 Gy (RR, 1,17; IC 95 %, 1,05–1,3; P = 0,01).
Consumo específico del antidepresivo paroxetina (RR, 1,29; IC 95 %, 1,08–1,54).
Los sobrevivientes que cumplieron las recomendaciones de los Centros para el Control y la Prevención de Enfermedades en relación con la actividad física vigorosa (RR, 0,90; IC 95 %, 0,82–0,97; P = 0,01) y que tenían un grado medio de ansiedad (RR, 0,86; IC 95 %, 0,75–0,99; P = 0,04) tuvieron un riesgo más bajo de obesidad.[129]
Alteraciones de la composición corporal posteriores a la leucemia linfoblástica aguda infantil
Evidencia (factores de riesgo de alteraciones en la composición corporal):
Las dosis moderadas de radiación craneal (18–24 Gy) administradas a los sobrevivientes de LLA se relacionan con obesidad; en particular en mujeres tratadas a una edad temprana.[120,122,130]
Las sobrevivientes de LLA infantil tratadas con dosis de radioterapia craneal de 24 Gy antes de los 5 años tienen 4 veces más probabilidades de ser obesas que aquellas mujeres no tratadas por cáncer.[120]
Además, las mujeres tratadas con dosis de 18 a 24 Gy de radioterapia craneal antes de los 10 años tienen un riesgo considerablemente mayor de aumento del IMC durante la juventud, en comparación con las mujeres tratadas por LLA con quimioterapia sola o aquellas de la población general.[122]
Estas mujeres también tienen una adiposidad visceral bastante mayor y, en consecuencia, resistencia a la insulina.[131,132]
Las alteraciones de la composición corporal se ven atenuadas en los varones.
No obstante, en un estudio de varones sobrevivientes a largo plazo de LLA (media de edad, 29 años), se observó una adiposidad corporal significativamente más alta que en los controles emparejados por edad, a pesar de tener peso e IMC normales.[133]
Los posibles indicadores de aumento de adiposidad son las concentraciones altas de leptina y concentraciones bajas de la globulina fijadora de hormonas sexuales.[133]
Los marcadores endocrinos testiculares séricos (testosterona, FSH o inhibina B) no se relacionaron con adiposidad corporal.[133]
Todos los regímenes terapéuticos para la LLA se relacionan con aumentos del IMC justo después de terminado el tratamiento y, quizás, con un riesgo más alto de obesidad a largo plazo.[123,124,134-136]
En varios estudios se notificó que los sobrevivientes de LLA tratados con quimioterapia sola también exhiben cambios a largo plazo en la composición corporal, con aumentos relativos de grasa corporal [132,137-139] y adiposidad visceral en comparación con masa magra.[131]
Estos cambios no se detectan si se usa solo el IMC para evaluar el riesgo metabólico en esta población.
Es posible que una predisposición genética al exceso de adiposidad se acentúe cuando el niño con LLA recibe irradiación craneal. Los determinantes genéticos del IMC identificados en la población general se investigaron en 2 grandes cohortes de sobrevivientes de cáncer infantil (n = 1458) (CCSS y SJLIFE).[140]
En los sobrevivientes de LLA tratados con radioterapia craneal en la niñez, los investigadores encontraron interacciones significativas entre el antecedente de radioterapia craneal en los sobrevivientes con variantes genéticas de la línea germinal que se sabe que afectan el riesgo genético basal de adiposidad. Esto indica que una predisposición genética a un exceso de adiposidad quizás se acentúe cuando la persona recibe radioterapia craneal para el tratamiento de una LLA infantil.
Evidencia (cambios de la composición corporal en adultos sobrevivientes de LLA infantil):
En un estudio de cohorte con 365 adultos sobrevivientes de LLA (149 tratados con radioterapia craneal y 216 tratados sin este tipo de radioterapia) se compararon la composición corporal, el equilibrio energético y el estado físico con personas emparejadas según la edad, el sexo y la raza.[141]
Las mujeres sobrevivientes que no estuvieron expuestas a irradiación craneal tuvieron valores de composición corporal comparables a los de sus pares.
Sin embargo, la circunferencia abdominal, la relación cintura-estatura y la masa adiposa total y porcentual fueron más altas en los varones y mujeres sobrevivientes expuestos a radiación craneal que entre los miembros del grupo de comparación.
Los sobrevivientes de ambos sexos expuestos a radioterapia craneal presentaron IMC y porcentaje de grasa corporal más altos que los sobrevivientes que no estuvieron expuestos a radioterapia craneal.
Si bien los sobrevivientes que no recibieron radioterapia craneal presentaron un equilibrio energético similar al de su grupo de comparación, tuvieron mediciones significativamente más altas de estado físico deficiente (deterioro de la flexibilidad, deficiencias sensitivomotoras periféricas, debilidad muscular proximal y tolerancia precaria al ejercicio).
En un estudio del CCSS, con base en mediciones de altura y peso autonotificados, se observó lo siguiente:
Los adultos sobrevivientes de LLA infantil tratados con quimioterapia sola no presentaron tasas significativamente más altas de obesidad que los hermanos del grupo de control.[120]
No hubo diferencias en los cambios del IMC entre estos grupos después de un período posterior de seguimiento promedio de 7,8 años.[122]
Investigadores suizos evaluaron el peso autonotificado en 1936 adultos sobrevivientes de LLA infantil, de linfoma no Hodgkin y de linfoma de Hodgkin (mediana de edad, 24 años; mediana de tiempo desde el diagnóstico, 17 años) y los compararon con hermanos y la población general.[142]
La proporción de sobrevivientes que notificaron sobrepeso (26 %) fue comparable a la de los hermanos (24 %) y la población suiza general (25 %).
No se obtuvo evidencia científica de una relación entre la dosis acumulada de glucocorticoides y el sobrepeso.
Además, tampoco se obtuvo evidencia que indicara que el uso de radioterapia craneal modificara el efecto de la dosis acumulada de glucocorticoides en el riesgo de sobrepeso.
Los resultados variables en los estudios seguramente guardan relación con el uso del IMC como parámetro para determinar una composición corporal anómala, que no evalúa de manera adecuada la adiposidad visceral que contribuye al riesgo metabólico en esta población.[143]
Alteraciones de la composición corporal posteriores al tratamiento de tumores del sistema nervioso central
Entre los sobrevivientes de tumores de encéfalo tratados con dosis más altas de radioterapia craneal, el riesgo más alto de presentar obesidad se observó en las mujeres tratadas a una edad más temprana.[144]
Los sobrevivientes de craneofaringioma tienen un riesgo bastante elevado de obesidad extrema debido a la ubicación del tumor y el daño hipotalámico que produce la resección quirúrgica.[145-148]
Se evaluó una cohorte de 661 sobrevivientes de tumores de encéfalo infantiles (tiempo medio de seguimiento, 7,3 años) para determinar el aumento de peso relacionado con la disfunción hipotálamo-hipofisaria. En esta cohorte se excluyó a los sobrevivientes que tenían craneofaringiomas o tumores hipofisarios.[149]
Entre los sobrevivientes de tumores de encéfalo infantiles con edades entre los 4 y los 20 años en el momento del seguimiento, el 20,3 % se clasificó como con sobrepeso y el 8,5 % como obeso, en comparación con el 10,5 % y el 2,7 %, respectivamente, en la población general neerlandesa de edad similar.
La disfunción hipotálamo-hipofisaria era más prevalente en los sobrevivientes con sobrepeso y obesidad que en los que tenían un peso normal.
El IMC estandarizado en el momento del diagnóstico, el diagnóstico de glioma de grado bajo y los antecedentes de diabetes insípida y pubertad precoz de origen central se relacionaron con aumento de peso, sobrepeso u obesidad.
Alteraciones de la composición corporal posteriores a un trasplante de células madre hematopoyéticas
Los sobrevivientes de cáncer infantil tratados con ICT en preparación para un TCMH alogénico tienen mediciones más altas de masa adiposa corporal (porcentaje de grasa) aunque su IMC suele ser normal.[113,150-152]
Entre los sobrevivientes de neoplasias hematológicas malignas tratados con TCMH alogénico, se observó un descenso longitudinal del IMC relacionado con una disminución importante de masa magra.[153]
Este hallazgo se atribuyó, en gran parte, al acondicionamiento para la ICT y la gravedad de la EICH crónica.[153]
Alteraciones en la composición corporal después de neoplasias sólidas
Los sobrevivientes de tumores abdominales y pélvicos sólidos que recibieron radioterapia están en riesgo de alteraciones en la composición corporal y en la salud metabólica que podrían afectar de manera adversa el desempeño funcional.
Entre 431 sobrevivientes de tumores abdominales o pélvicos infantiles (mediana de edad alcanzada, 29,9 años), las dosis acumuladas más altas de radioterapia abdominal o pélvica se asociaron con menor masa muscular magra en hombres y mujeres.[154]
En comparación con los sobrevivientes que tienen una masa magra normal (alta) y masa grasa normal (baja), los sobrevivientes con masa magra normal y masa grasa alta presentaron un riesgo significativamente más alto de resistencia a la insulina, colesterol HDL bajo y fuerza del cuádriceps reducida, así como menor distancia durante una caminata de 6 minutos.[154]
Composición corporal y fragilidad
Los adultos jóvenes sobrevivientes de cáncer infantil tienen una prevalencia más alta de la esperada de fragilidad, un fenotipo caracterizado por una masa muscular baja, agotamiento autonotificado, bajo gasto energético, velocidad lenta de marcha y debilidad.[155]
El estado de estas personas se considera prefrágil si tienen 2 de estas 5 características y frágil si tienen 3 o más de estas características.[155]
El fenotipo de fragilidad aumenta su prevalencia con el envejecimiento y se relacionó con un exceso de riesgo de mortalidad y afecciones crónicas.[155]
En los participantes del CCSS, la prevalencia general de la fragilidad entre los sobrevivientes (6,4 %) fue 3 veces más alta que la notificada por los hermanos del grupo de control (2,2 %).[156]
La prevalencia más alta de la fragilidad se notificó en sobrevivientes de tumores del SNC (9,5 %) y tumores de hueso (8,1 %).
La radiación craneal, la radiación pélvica (≥34 Gy) y la cirugía de pulmón fueron factores de riesgo independientes de fragilidad en la cohorte del CCSS.
En los participantes del estudio SJLIFE (mediana de edad en el momento de ingresar al estudio, 30 años), la prevalencia de fragilidad aumentó del 6,2 % al 13,6 % durante los 5 años de seguimiento.[157]
Los factores de riesgo de fragilidad en el momento del seguimiento fueron la radioterapia torácica con dosis de 20 Gy o más altas (OR, 1,98), las afecciones cardíacas (OR, 1,58) y neurológicas (OR, 2,58), la falta de entrenamiento de fortalecimiento (OR, 1,74), el estilo de vida sedentario (OR, 1,75), y la fragilidad en el momento de ingresar al estudio (OR, 11,12).
El antecedente de fragilidad fue el factor de riesgo más fuerte para la muerte durante el seguimiento (OR, 3,52).
En un análisis del estudio SJLIFE (intervalo de seguimiento, 5 años), los adultos jóvenes sobrevivientes de cáncer infantil en estado prefrágil o frágil (n = 205) presentaron reducciones significativas en los dominios neurocognitivos que suelen relacionarse con demencia y enfermedad de Alzheimer.[158]
En los sobrevivientes frágiles se redujo más la puntuación de memoria verbal a corto plazo y el funcionamiento ejecutivo que en los sobrevivientes sin fragilidad.
Los sobrevivientes frágiles y prefrágiles experimentaron reducciones significativas en la atención enfocada, en comparación con los sobrevivientes sin fragilidad.
La prevalencia y los factores de riesgo de prefragilidad, fragilidad y sarcopenia se evaluaron en un estudio transversal DCCSS-LATER de adultos sobrevivientes (media de edad, 33,1 años en el momento de la participación) de cáncer infantil que se trataron entre 1963 y 2001.[159]
La tasa de prefragilidad fue del 20,3 %, la tasa de fragilidad fue del 7,4 % y la tasa de sarcopenia fue del 4,4 %.
Los siguientes son importantes factores de riesgo de prefragilidad:
Peso insuficiente (OR, 3,38; IC 95 %, 1,92–5,95).
Obesidad (OR, 1,67; IC 95 %, 1,14–2,43).
Exposición a irradiación craneal (OR, 2,07; IC 95 %, 1,47–2,93).
Exposición a ITC (OR, 3,17; IC 95 %, 1,77–5,70).
Exposición a dosis de cisplatino de al menos 600 mg/m2 (OR, 3,75; IC 95 %, 1,82–7,74).
Deficiencia de la hormona de crecimiento (OR, 2,25; IC 95 %, 1,23–4,09).
Hipertiroidismo (OR, 3,72; IC 95 %, 1,63–8,47).
Baja densidad mineral ósea (OR para el puntaje Z ≤−1 y >-2, 1,80; IC 95 %, 1,31–2,47 y OR para el puntaje Z ≤-2, 3,37; IC 95 %, 2,20–5,15).
Deficiencia de ácido fólico (OR, 1,87; IC 95 %, 1,31–2,68).
Los siguientes son importantes factores de riesgo de sarcopenia:
Sexo masculino (OR, 4,56; IC 95 %, 2,26–9,17).
Bajo IMC (constante, OR, 0,52; IC 95 %, 0,45–0,60).
Exposición a irradiación craneal (OR, 3,87; IC 95 %, 1,80–8,31).
Exposición a ITC (OR, 4,52; IC 95 %, 1,67–12,20).
Hipogonadismo (OR, 3,96; IC 95 %, 1,40–11,18).
Deficiencia de la hormona de crecimiento (OR, 4,66; IC 95 %, 1,44–15,15).
Deficiencia de vitamina B12 (OR, 6,26).
Es posible que los sobrevivientes de cáncer infantil experimenten un envejecimiento biológico acelerado, que se traduce en fragilidad y muerte prematuras. Los investigadores usaron la cohorte SJLIFE para evaluar un panel de los biomarcadores de envejecimiento más recientes, más estudiados y con la mejor evidencia. En su análisis incluyeron 7 medidas de aceleración de la edad biológica. Los adultos sobrevivientes de cáncer infantil de la cohorte SJLIFE se compararon con otros 2 grupos de control (controles comunitarios y de la National Health and Nutrition Examination Survey). El objetivo era determinar si los sobrevivientes de cáncer infantil (n = 4117) eran biológicamente mayores y envejecían de forma más rápida que los controles que no habían tenido cáncer infantil.[160]
Los sobrevivientes de cáncer envejecieron un 5 % más rápido cada año y eran, en promedio, entre 0,6 y 6,44 años mayores biológicamente que los controles comunitarios.
Los sobrevivientes eran biológicamente de 5 a 16 años mayores de lo esperado, en comparación con adultos de su edad cronológica en el nivel de población.
Los sobrevivientes que recibieron tratamiento con TCMH y quimioterapia con alcaloides de vinca mostraron las trayectorias más rápidas de envejecimiento biológico.
Los sobrevivientes biológicamente mayores y con envejecimiento más rápido tuvieron, de manera consistente y sólida, un mayor riesgo de debilidad y murieron más temprano que aquellos con un envejecimiento biológico más lento.
El objetivo de la investigación en curso es dilucidar las características fisiopatológicas de la fragilidad, y formular y poner a prueba intervenciones para prevenir o revertir esta afección.
En el Cuadro 13 se resumen los efectos tardíos en la composición corporal y los exámenes médicos de detección correspondientes.
Cuadro 13. Efectos tardíos en la composición corporala
Brignardello E, Felicetti F, Castiglione A, et al.: Endocrine health conditions in adult survivors of childhood cancer: the need for specialized adult-focused follow-up clinics. Eur J Endocrinol 168 (3): 465-72, 2013. [PUBMED Abstract]
Mostoufi-Moab S, Seidel K, Leisenring WM, et al.: Endocrine Abnormalities in Aging Survivors of Childhood Cancer: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 34 (27): 3240-7, 2016. [PUBMED Abstract]
Clement SC, Schouten-van Meeteren AY, Boot AM, et al.: Prevalence and Risk Factors of Early Endocrine Disorders in Childhood Brain Tumor Survivors: A Nationwide, Multicenter Study. J Clin Oncol 34 (36): 4362-4370, 2016. [PUBMED Abstract]
Chemaitilly W, Li Z, Huang S, et al.: Anterior hypopituitarism in adult survivors of childhood cancers treated with cranial radiotherapy: a report from the St Jude Lifetime Cohort study. J Clin Oncol 33 (5): 492-500, 2015. [PUBMED Abstract]
González Briceño LG, Kariyawasam D, Samara-Boustani D, et al.: High Prevalence of Early Endocrine Disorders After Childhood Brain Tumors in a Large Cohort. J Clin Endocrinol Metab 107 (5): e2156-e2166, 2022. [PUBMED Abstract]
Clement SC, van Rijn RR, van Eck-Smit BL, et al.: Long-term efficacy of current thyroid prophylaxis and future perspectives on thyroid protection during 131I-metaiodobenzylguanidine treatment in children with neuroblastoma. Eur J Nucl Med Mol Imaging 42 (5): 706-15, 2015. [PUBMED Abstract]
Clement SC, van Eck-Smit BL, van Trotsenburg AS, et al.: Long-term follow-up of the thyroid gland after treatment with 131I-Metaiodobenzylguanidine in children with neuroblastoma: importance of continuous surveillance. Pediatr Blood Cancer 60 (11): 1833-8, 2013. [PUBMED Abstract]
Bombardieri E, Giammarile F, Aktolun C, et al.: 131I/123I-metaiodobenzylguanidine (mIBG) scintigraphy: procedure guidelines for tumour imaging. Eur J Nucl Med Mol Imaging 37 (12): 2436-46, 2010. [PUBMED Abstract]
Sharp SE, Trout AT, Weiss BD, et al.: MIBG in Neuroblastoma Diagnostic Imaging and Therapy. Radiographics 36 (1): 258-78, 2016 Jan-Feb. [PUBMED Abstract]
Clement SC, Tytgat GAM, van Trotsenburg ASP, et al.: Thyroid function after diagnostic 123I-metaiodobenzylguanidine in children with neuroblastic tumors. Ann Nucl Med 36 (6): 579-585, 2022. [PUBMED Abstract]
Verloop H, Louwerens M, Schoones JW, et al.: Risk of hypothyroidism following hemithyroidectomy: systematic review and meta-analysis of prognostic studies. J Clin Endocrinol Metab 97 (7): 2243-55, 2012. [PUBMED Abstract]
Zatelli MC, Lamartina L, Meringolo D, et al.: Thyroid nodule recurrence following lobo-isthmectomy: incidence, patient's characteristics, and risk factors. J Endocrinol Invest 41 (12): 1469-1475, 2018. [PUBMED Abstract]
Chemaitilly W, Li Z, Brinkman TM, et al.: Primary hypothyroidism in childhood cancer survivors: Prevalence, risk factors, and long-term consequences. Cancer 128 (3): 606-614, 2022. [PUBMED Abstract]
Bölling T, Geisenheiser A, Pape H, et al.: Hypothyroidism after head-and-neck radiotherapy in children and adolescents: preliminary results of the "Registry for the Evaluation of Side Effects After Radiotherapy in Childhood and Adolescence" (RiSK). Int J Radiat Oncol Biol Phys 81 (5): e787-91, 2011. [PUBMED Abstract]
Inskip PD, Veiga LHS, Brenner AV, et al.: Hypothyroidism after Radiation Therapy for Childhood Cancer: A Report from the Childhood Cancer Survivor Study. Radiat Res 190 (2): 117-132, 2018. [PUBMED Abstract]
Sklar C, Whitton J, Mertens A, et al.: Abnormalities of the thyroid in survivors of Hodgkin's disease: data from the Childhood Cancer Survivor Study. J Clin Endocrinol Metab 85 (9): 3227-32, 2000. [PUBMED Abstract]
Vatner RE, Niemierko A, Misra M, et al.: Endocrine Deficiency As a Function of Radiation Dose to the Hypothalamus and Pituitary in Pediatric and Young Adult Patients With Brain Tumors. J Clin Oncol 36 (28): 2854-2862, 2018. [PUBMED Abstract]
Aldrich KD, Horne VE, Bielamowicz K, et al.: Comparison of hypothyroidism, growth hormone deficiency, and adrenal insufficiency following proton and photon radiotherapy in children with medulloblastoma. J Neurooncol 155 (1): 93-100, 2021. [PUBMED Abstract]
Milano MT, Vargo JA, Yorke ED, et al.: Primary Hypothyroidism in Childhood Cancer Survivors Treated With Radiation Therapy: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 482-493, 2024. [PUBMED Abstract]
Wheeler G, Grassberger C, Samers J, et al.: Central Endocrine Complications Among Childhood Cancer Survivors Treated With Radiation Therapy: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 457-466, 2024. [PUBMED Abstract]
Gleeson HK, Darzy K, Shalet SM: Late endocrine, metabolic and skeletal sequelae following treatment of childhood cancer. Best Pract Res Clin Endocrinol Metab 16 (2): 335-48, 2002. [PUBMED Abstract]
Inskip PD, Veiga LHS, Brenner AV, et al.: Hyperthyroidism After Radiation Therapy for Childhood Cancer: A Report from the Childhood Cancer Survivor Study. Int J Radiat Oncol Biol Phys 104 (2): 415-424, 2019. [PUBMED Abstract]
Clausen CT, Hasle H, Holmqvist AS, et al.: Hyperthyroidism as a late effect in childhood cancer survivors - an Adult Life after Childhood Cancer in Scandinavia (ALiCCS) study. Acta Oncol 58 (2): 227-231, 2019. [PUBMED Abstract]
Bhatti P, Veiga LH, Ronckers CM, et al.: Risk of second primary thyroid cancer after radiotherapy for a childhood cancer in a large cohort study: an update from the childhood cancer survivor study. Radiat Res 174 (6): 741-52, 2010. [PUBMED Abstract]
Haddy N, El-Fayech C, Guibout C, et al.: Thyroid adenomas after solid cancer in childhood. Int J Radiat Oncol Biol Phys 84 (2): e209-15, 2012. [PUBMED Abstract]
Ronckers CM, Sigurdson AJ, Stovall M, et al.: Thyroid cancer in childhood cancer survivors: a detailed evaluation of radiation dose response and its modifiers. Radiat Res 166 (4): 618-28, 2006. [PUBMED Abstract]
Sigurdson AJ, Ronckers CM, Mertens AC, et al.: Primary thyroid cancer after a first tumour in childhood (the Childhood Cancer Survivor Study): a nested case-control study. Lancet 365 (9476): 2014-23, 2005 Jun 11-17. [PUBMED Abstract]
van Santen HM, Tytgat GA, van de Wetering MD, et al.: Differentiated thyroid carcinoma after 131I-MIBG treatment for neuroblastoma during childhood: description of the first two cases. Thyroid 22 (6): 643-6, 2012. [PUBMED Abstract]
Veiga LH, Lubin JH, Anderson H, et al.: A pooled analysis of thyroid cancer incidence following radiotherapy for childhood cancer. Radiat Res 178 (4): 365-76, 2012. [PUBMED Abstract]
Kovalchik SA, Ronckers CM, Veiga LH, et al.: Absolute risk prediction of second primary thyroid cancer among 5-year survivors of childhood cancer. J Clin Oncol 31 (1): 119-27, 2013. [PUBMED Abstract]
Vivanco M, Dalle JH, Alberti C, et al.: Malignant and benign thyroid nodules after total body irradiation preceding hematopoietic cell transplantation during childhood. Eur J Endocrinol 167 (2): 225-33, 2012. [PUBMED Abstract]
Li Z, Franklin J, Zelcer S, et al.: Ultrasound surveillance for thyroid malignancies in survivors of childhood cancer following radiotherapy: a single institutional experience. Thyroid 24 (12): 1796-805, 2014. [PUBMED Abstract]
Brignardello E, Felicetti F, Castiglione A, et al.: Ultrasound surveillance for radiation-induced thyroid carcinoma in adult survivors of childhood cancer. Eur J Cancer 55: 74-80, 2016. [PUBMED Abstract]
Clement SC, Kremer LCM, Verburg FA, et al.: Balancing the benefits and harms of thyroid cancer surveillance in survivors of Childhood, adolescent and young adult cancer: Recommendations from the international Late Effects of Childhood Cancer Guideline Harmonization Group in collaboration with the PanCareSurFup Consortium. Cancer Treat Rev 63: 28-39, 2018. [PUBMED Abstract]
Baran JA, Halada S, Bauer AJ, et al.: Thyroid Ultrasound Screening in Childhood Cancer Survivors following Radiotherapy. Horm Res Paediatr 97 (3): 243-253, 2024. [PUBMED Abstract]
Metzger ML, Howard SC, Hudson MM, et al.: Natural history of thyroid nodules in survivors of pediatric Hodgkin lymphoma. Pediatr Blood Cancer 46 (3): 314-9, 2006. [PUBMED Abstract]
Francis GL, Waguespack SG, Bauer AJ, et al.: Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 25 (7): 716-59, 2015. [PUBMED Abstract]
Sanders JE, Hoffmeister PA, Woolfrey AE, et al.: Thyroid function following hematopoietic cell transplantation in children: 30 years' experience. Blood 113 (2): 306-8, 2009. [PUBMED Abstract]
Cattoni A, Molinari S, Gaiero A, et al.: Thyroid Disorders Following Hematopoietic Stem Cell Transplantation in Childhood: Impact of Conditioning Regimen on Thyroid Dysfunction, Volume Changes, and Occurrence of Nodules. Transplant Cell Ther 28 (8): 506.e1-506.e12, 2022. [PUBMED Abstract]
van Iersel L, Mulder RL, Denzer C, et al.: Hypothalamic-Pituitary and Other Endocrine Surveillance Among Childhood Cancer Survivors. Endocr Rev 43 (5): 794-823, 2022. [PUBMED Abstract]
Gurney JG, Kadan-Lottick NS, Packer RJ, et al.: Endocrine and cardiovascular late effects among adult survivors of childhood brain tumors: Childhood Cancer Survivor Study. Cancer 97 (3): 663-73, 2003. [PUBMED Abstract]
Hudson MM, Ness KK, Gurney JG, et al.: Clinical ascertainment of health outcomes among adults treated for childhood cancer. JAMA 309 (22): 2371-81, 2013. [PUBMED Abstract]
Sklar CA, Antal Z, Chemaitilly W, et al.: Hypothalamic-Pituitary and Growth Disorders in Survivors of Childhood Cancer: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 103 (8): 2761-2784, 2018. [PUBMED Abstract]
Ramelli GP, von der Weid N, Stanga Z, et al.: Suprasellar germinomas in childhood and adolescence: diagnostic pitfalls. J Pediatr Endocrinol Metab 11 (6): 693-7, 1998 Nov-Dec. [PUBMED Abstract]
Vinchon M, Baroncini M, Leblond P, et al.: Morbidity and tumor-related mortality among adult survivors of pediatric brain tumors: a review. Childs Nerv Syst 27 (5): 697-704, 2011. [PUBMED Abstract]
Fahrner B, Prosch H, Minkov M, et al.: Long-term outcome of hypothalamic pituitary tumors in Langerhans cell histiocytosis. Pediatr Blood Cancer 58 (4): 606-10, 2012. [PUBMED Abstract]
Lawson SA, Horne VE, Golekoh MC, et al.: Hypothalamic-pituitary function following childhood brain tumors: Analysis of prospective annual endocrine screening. Pediatr Blood Cancer 66 (5): e27631, 2019. [PUBMED Abstract]
Lebbink CA, Ringers TP, Schouten-van Meeteren AYN, et al.: Prevalence and risk factors of hypothalamic-pituitary dysfunction in infant and toddler childhood brain tumor survivors. Eur J Endocrinol 185 (4): 597-606, 2021. [PUBMED Abstract]
Fraser O, Crowne E, Tacey M, et al.: Correlating measured radiotherapy dose with patterns of endocrinopathy: The importance of minimizing pituitary dose. Pediatr Blood Cancer 69 (11): e29847, 2022. [PUBMED Abstract]
van Iersel L, Li Z, Srivastava DK, et al.: Hypothalamic-Pituitary Disorders in Childhood Cancer Survivors: Prevalence, Risk Factors and Long-Term Health Outcomes. J Clin Endocrinol Metab 104 (12): 6101-6115, 2019. [PUBMED Abstract]
Viana MB, Vilela MI: Height deficit during and many years after treatment for acute lymphoblastic leukemia in children: a review. Pediatr Blood Cancer 50 (2 Suppl): 509-16; discussion 517, 2008. [PUBMED Abstract]
Merchant TE, Goloubeva O, Pritchard DL, et al.: Radiation dose-volume effects on growth hormone secretion. Int J Radiat Oncol Biol Phys 52 (5): 1264-70, 2002. [PUBMED Abstract]
Merchant TE, Rose SR, Bosley C, et al.: Growth hormone secretion after conformal radiation therapy in pediatric patients with localized brain tumors. J Clin Oncol 29 (36): 4776-80, 2011. [PUBMED Abstract]
Merchant TE, Wu S, Onar-Thomas A, et al.: Endocrinopathy After Treatment for Medulloblastoma: Results From the SJMB03 Trial of Risk-Adapted Radiation Therapy. Int J Radiat Oncol Biol Phys 116 (3): 560-568, 2023. [PUBMED Abstract]
van Iersel L, van Santen HM, Potter B, et al.: Clinical impact of hypothalamic-pituitary disorders after conformal radiation therapy for pediatric low-grade glioma or ependymoma. Pediatr Blood Cancer 67 (12): e28723, 2020. [PUBMED Abstract]
Sklar C, Mertens A, Walter A, et al.: Final height after treatment for childhood acute lymphoblastic leukemia: comparison of no cranial irradiation with 1800 and 2400 centigrays of cranial irradiation. J Pediatr 123 (1): 59-64, 1993. [PUBMED Abstract]
Chow EJ, Friedman DL, Yasui Y, et al.: Decreased adult height in survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. J Pediatr 150 (4): 370-5, 375.e1, 2007. [PUBMED Abstract]
Vandecruys E, Dhooge C, Craen M, et al.: Longitudinal linear growth and final height is impaired in childhood acute lymphoblastic leukemia survivors after treatment without cranial irradiation. J Pediatr 163 (1): 268-73, 2013. [PUBMED Abstract]
Browne EK, Zhou Y, Chemaitilly W, et al.: Changes in body mass index, height, and weight in children during and after therapy for acute lymphoblastic leukemia. Cancer 124 (21): 4248-4259, 2018. [PUBMED Abstract]
Demoor-Goldschmidt C, Allodji RS, Journy N, et al.: Risk Factors for Small Adult Height in Childhood Cancer Survivors. J Clin Oncol 38 (16): 1785-1796, 2020. [PUBMED Abstract]
Mizumoto M, Oshiro Y, Pan H, et al.: Height after photon craniospinal irradiation in pediatric patients treated for central nervous system embryonal tumors. Pediatr Blood Cancer 67 (10): e28617, 2020. [PUBMED Abstract]
Ogilvy-Stuart AL, Clark DJ, Wallace WH, et al.: Endocrine deficit after fractionated total body irradiation. Arch Dis Child 67 (9): 1107-10, 1992. [PUBMED Abstract]
Willi SM, Cooke K, Goldwein J, et al.: Growth in children after bone marrow transplantation for advanced neuroblastoma compared with growth after transplantation for leukemia or aplastic anemia. J Pediatr 120 (5): 726-32, 1992. [PUBMED Abstract]
Wingard JR, Plotnick LP, Freemer CS, et al.: Growth in children after bone marrow transplantation: busulfan plus cyclophosphamide versus cyclophosphamide plus total body irradiation. Blood 79 (4): 1068-73, 1992. [PUBMED Abstract]
Huma Z, Boulad F, Black P, et al.: Growth in children after bone marrow transplantation for acute leukemia. Blood 86 (2): 819-24, 1995. [PUBMED Abstract]
Bernard F, Bordigoni P, Simeoni MC, et al.: Height growth during adolescence and final height after haematopoietic SCT for childhood acute leukaemia: the impact of a conditioning regimen with BU or TBI. Bone Marrow Transplant 43 (8): 637-42, 2009. [PUBMED Abstract]
Chemaitilly W, Sklar CA: Endocrine complications of hematopoietic stem cell transplantation. Endocrinol Metab Clin North Am 36 (4): 983-98; ix, 2007. [PUBMED Abstract]
Socié G, Salooja N, Cohen A, et al.: Nonmalignant late effects after allogeneic stem cell transplantation. Blood 101 (9): 3373-85, 2003. [PUBMED Abstract]
Cohen A, Rovelli A, Bakker B, et al.: Final height of patients who underwent bone marrow transplantation for hematological disorders during childhood: a study by the Working Party for Late Effects-EBMT. Blood 93 (12): 4109-15, 1999. [PUBMED Abstract]
Brennan BM, Shalet SM: Endocrine late effects after bone marrow transplant. Br J Haematol 118 (1): 58-66, 2002. [PUBMED Abstract]
Sfeir JG, Kittah NEN, Tamhane SU, et al.: Diagnosis of GH Deficiency as a Late Effect of Radiotherapy in Survivors of Childhood Cancers. J Clin Endocrinol Metab 103 (8): 2785-2793, 2018. [PUBMED Abstract]
Raman S, Grimberg A, Waguespack SG, et al.: Risk of Neoplasia in Pediatric Patients Receiving Growth Hormone Therapy--A Report From the Pediatric Endocrine Society Drug and Therapeutics Committee. J Clin Endocrinol Metab 100 (6): 2192-203, 2015. [PUBMED Abstract]
Chemaitilly W, Robison LL: Safety of growth hormone treatment in patients previously treated for cancer. Endocrinol Metab Clin North Am 41 (4): 785-92, 2012. [PUBMED Abstract]
Sklar CA, Mertens AC, Mitby P, et al.: Risk of disease recurrence and second neoplasms in survivors of childhood cancer treated with growth hormone: a report from the Childhood Cancer Survivor Study. J Clin Endocrinol Metab 87 (7): 3136-41, 2002. [PUBMED Abstract]
Ergun-Longmire B, Mertens AC, Mitby P, et al.: Growth hormone treatment and risk of second neoplasms in the childhood cancer survivor. J Clin Endocrinol Metab 91 (9): 3494-8, 2006. [PUBMED Abstract]
Patterson BC, Chen Y, Sklar CA, et al.: Growth hormone exposure as a risk factor for the development of subsequent neoplasms of the central nervous system: a report from the childhood cancer survivor study. J Clin Endocrinol Metab 99 (6): 2030-7, 2014. [PUBMED Abstract]
Bogarin R, Steinbok P: Growth hormone treatment and risk of recurrence or progression of brain tumors in children: a review. Childs Nerv Syst 25 (3): 273-9, 2009. [PUBMED Abstract]
Tamhane S, Sfeir JG, Kittah NEN, et al.: GH Therapy in Childhood Cancer Survivors: A Systematic Review and Meta-Analysis. J Clin Endocrinol Metab 103 (8): 2794-2801, 2018. [PUBMED Abstract]
Thomas-Teinturier C, Oliver-Petit I, Pacquement H, et al.: Influence of growth hormone therapy on the occurrence of a second neoplasm in survivors of childhood cancer. Eur J Endocrinol 183 (4): 471-480, 2020. [PUBMED Abstract]
Boguszewski MCS, Boguszewski CL, Chemaitilly W, et al.: Safety of growth hormone replacement in survivors of cancer and intracranial and pituitary tumours: a consensus statement. Eur J Endocrinol 186 (6): P35-P52, 2022. [PUBMED Abstract]
Chemaitilly W, Merchant TE, Li Z, et al.: Central precocious puberty following the diagnosis and treatment of paediatric cancer and central nervous system tumours: presentation and long-term outcomes. Clin Endocrinol (Oxf) 84 (3): 361-71, 2016. [PUBMED Abstract]
Armstrong GT, Whitton JA, Gajjar A, et al.: Abnormal timing of menarche in survivors of central nervous system tumors: A report from the Childhood Cancer Survivor Study. Cancer 115 (11): 2562-70, 2009. [PUBMED Abstract]
Didcock E, Davies HA, Didi M, et al.: Pubertal growth in young adult survivors of childhood leukemia. J Clin Oncol 13 (10): 2503-7, 1995. [PUBMED Abstract]
Gan HW, Phipps K, Aquilina K, et al.: Neuroendocrine Morbidity After Pediatric Optic Gliomas: A Longitudinal Analysis of 166 Children Over 30 Years. J Clin Endocrinol Metab 100 (10): 3787-99, 2015. [PUBMED Abstract]
Chemaitilly W, Sklar CA: Endocrine complications in long-term survivors of childhood cancers. Endocr Relat Cancer 17 (3): R141-59, 2010. [PUBMED Abstract]
Greulich WW, Pyle SI: Radiographic Atlas of Skeletal Development of Hand and Wrist. 2nd ed. Stanford University Press, 1959.
Gleeson HK, Stoeter R, Ogilvy-Stuart AL, et al.: Improvements in final height over 25 years in growth hormone (GH)-deficient childhood survivors of brain tumors receiving GH replacement. J Clin Endocrinol Metab 88 (8): 3682-9, 2003. [PUBMED Abstract]
Laughton SJ, Merchant TE, Sklar CA, et al.: Endocrine outcomes for children with embryonal brain tumors after risk-adapted craniospinal and conformal primary-site irradiation and high-dose chemotherapy with stem-cell rescue on the SJMB-96 trial. J Clin Oncol 26 (7): 1112-8, 2008. [PUBMED Abstract]
Bielamowicz K, Okcu MF, Sonabend R, et al.: Hypothyroidism after craniospinal irradiation with proton or photon therapy in patients with medulloblastoma. Pediatr Hematol Oncol 35 (4): 257-267, 2018. [PUBMED Abstract]
van Iersel L, Xu J, Potter BS, et al.: Clinical Importance of Free Thyroxine Concentration Decline After Radiotherapy for Pediatric and Adolescent Brain Tumors. J Clin Endocrinol Metab 104 (11): 4998-5007, 2019. [PUBMED Abstract]
Kazlauskaite R, Evans AT, Villabona CV, et al.: Corticotropin tests for hypothalamic-pituitary- adrenal insufficiency: a metaanalysis. J Clin Endocrinol Metab 93 (11): 4245-53, 2008. [PUBMED Abstract]
Rose SR, Danish RK, Kearney NS, et al.: ACTH deficiency in childhood cancer survivors. Pediatr Blood Cancer 45 (6): 808-13, 2005. [PUBMED Abstract]
Clement SC, Peeters RP, Ronckers CM, et al.: Intermediate and long-term adverse effects of radioiodine therapy for differentiated thyroid carcinoma--a systematic review. Cancer Treat Rev 41 (10): 925-34, 2015. [PUBMED Abstract]
Follin C, Erfurth EM: Long-Term Effect of Cranial Radiotherapy on Pituitary-Hypothalamus Area in Childhood Acute Lymphoblastic Leukemia Survivors. Curr Treat Options Oncol 17 (9): 50, 2016. [PUBMED Abstract]
Rensen N, Gemke RJ, van Dalen EC, et al.: Hypothalamic-pituitary-adrenal (HPA) axis suppression after treatment with glucocorticoid therapy for childhood acute lymphoblastic leukaemia. Cochrane Database Syst Rev 11 (11): CD008727, 2017. [PUBMED Abstract]
Constine LS, Woolf PD, Cann D, et al.: Hypothalamic-pituitary dysfunction after radiation for brain tumors. N Engl J Med 328 (2): 87-94, 1993. [PUBMED Abstract]
Constine LS, Rubin P, Woolf PD, et al.: Hyperprolactinemia and hypothyroidism following cytotoxic therapy for central nervous system malignancies. J Clin Oncol 5 (11): 1841-51, 1987. [PUBMED Abstract]
Melmed S, Casanueva FF, Hoffman AR, et al.: Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 96 (2): 273-88, 2011. [PUBMED Abstract]
National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III): Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 106 (25): 3143-421, 2002. [PUBMED Abstract]
Bolier M, de Winter DTC, Pluimakers VG, et al.: Prevalence and determinants of metabolic syndrome in 2338 childhood cancer survivors: A Dutch Childhood Cancer Survivor LATER 2 study. Cancer 131 (1): e35681, 2025. [PUBMED Abstract]
Saultier P, Auquier P, Bertrand Y, et al.: Metabolic syndrome in long-term survivors of childhood acute leukemia treated without hematopoietic stem cell transplantation: an L.E.A. study. Haematologica 101 (12): 1603-1610, 2016. [PUBMED Abstract]
Nottage KA, Ness KK, Li C, et al.: Metabolic syndrome and cardiovascular risk among long-term survivors of acute lymphoblastic leukaemia - From the St. Jude Lifetime Cohort. Br J Haematol 165 (3): 364-74, 2014. [PUBMED Abstract]
Oudin C, Berbis J, Bertrand Y, et al.: Prevalence and characteristics of metabolic syndrome in adults from the French childhood leukemia survivors' cohort: a comparison with controls from the French population. Haematologica 103 (4): 645-654, 2018. [PUBMED Abstract]
van Waas M, Neggers SJ, Raat H, et al.: Abdominal radiotherapy: a major determinant of metabolic syndrome in nephroblastoma and neuroblastoma survivors. PLoS One 7 (12): e52237, 2012. [PUBMED Abstract]
Smith WA, Li C, Nottage KA, et al.: Lifestyle and metabolic syndrome in adult survivors of childhood cancer: a report from the St. Jude Lifetime Cohort Study. Cancer 120 (17): 2742-50, 2014. [PUBMED Abstract]
Jones LW, Liu Q, Armstrong GT, et al.: Exercise and risk of major cardiovascular events in adult survivors of childhood hodgkin lymphoma: a report from the childhood cancer survivor study. J Clin Oncol 32 (32): 3643-50, 2014. [PUBMED Abstract]
Scott JM, Li N, Liu Q, et al.: Association of Exercise With Mortality in Adult Survivors of Childhood Cancer. JAMA Oncol 4 (10): 1352-1358, 2018. [PUBMED Abstract]
Baker KS, Ness KK, Steinberger J, et al.: Diabetes, hypertension, and cardiovascular events in survivors of hematopoietic cell transplantation: a report from the bone marrow transplantation survivor study. Blood 109 (4): 1765-72, 2007. [PUBMED Abstract]
van Waas M, Neggers SJ, Uitterlinden AG, et al.: Treatment factors rather than genetic variation determine metabolic syndrome in childhood cancer survivors. Eur J Cancer 49 (3): 668-75, 2013. [PUBMED Abstract]
de Vathaire F, El-Fayech C, Ben Ayed FF, et al.: Radiation dose to the pancreas and risk of diabetes mellitus in childhood cancer survivors: a retrospective cohort study. Lancet Oncol 13 (10): 1002-10, 2012. [PUBMED Abstract]
Armenian SH, Chemaitilly W, Chen M, et al.: National Institutes of Health Hematopoietic Cell Transplantation Late Effects Initiative: The Cardiovascular Disease and Associated Risk Factors Working Group Report. Biol Blood Marrow Transplant 23 (2): 201-210, 2017. [PUBMED Abstract]
Lega IC, Pole JD, Austin PC, et al.: Diabetes Risk in Childhood Cancer Survivors: A Population-Based Study. Can J Diabetes 42 (5): 533-539, 2018. [PUBMED Abstract]
Baker KS, Chow EJ, Goodman PJ, et al.: Impact of treatment exposures on cardiovascular risk and insulin resistance in childhood cancer survivors. Cancer Epidemiol Biomarkers Prev 22 (11): 1954-63, 2013. [PUBMED Abstract]
Meacham LR, Chow EJ, Ness KK, et al.: Cardiovascular risk factors in adult survivors of pediatric cancer--a report from the childhood cancer survivor study. Cancer Epidemiol Biomarkers Prev 19 (1): 170-81, 2010. [PUBMED Abstract]
van Santen HM, Geskus RB, Raemaekers S, et al.: Changes in body mass index in long-term childhood cancer survivors. Cancer 121 (23): 4197-204, 2015. [PUBMED Abstract]
Meacham LR, Gurney JG, Mertens AC, et al.: Body mass index in long-term adult survivors of childhood cancer: a report of the Childhood Cancer Survivor Study. Cancer 103 (8): 1730-9, 2005. [PUBMED Abstract]
Tonorezos ES, Chou JF, Moskowitz CS, et al.: Risk of increased mortality in underweight survivors: A brief report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer 71 (8): e31080, 2024. [PUBMED Abstract]
Oeffinger KC, Mertens AC, Sklar CA, et al.: Obesity in adult survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. J Clin Oncol 21 (7): 1359-65, 2003. [PUBMED Abstract]
Sklar CA, Mertens AC, Walter A, et al.: Changes in body mass index and prevalence of overweight in survivors of childhood acute lymphoblastic leukemia: role of cranial irradiation. Med Pediatr Oncol 35 (2): 91-5, 2000. [PUBMED Abstract]
Garmey EG, Liu Q, Sklar CA, et al.: Longitudinal changes in obesity and body mass index among adult survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. J Clin Oncol 26 (28): 4639-45, 2008. [PUBMED Abstract]
Chow EJ, Pihoker C, Hunt K, et al.: Obesity and hypertension among children after treatment for acute lymphoblastic leukemia. Cancer 110 (10): 2313-20, 2007. [PUBMED Abstract]
Withycombe JS, Post-White JE, Meza JL, et al.: Weight patterns in children with higher risk ALL: A report from the Children's Oncology Group (COG) for CCG 1961. Pediatr Blood Cancer 53 (7): 1249-54, 2009. [PUBMED Abstract]
Nathan PC, Jovcevska V, Ness KK, et al.: The prevalence of overweight and obesity in pediatric survivors of cancer. J Pediatr 149 (4): 518-25, 2006. [PUBMED Abstract]
Oeffinger KC: Are survivors of acute lymphoblastic leukemia (ALL) at increased risk of cardiovascular disease? Pediatr Blood Cancer 50 (2 Suppl): 462-7; discussion 468, 2008. [PUBMED Abstract]
Brennan BM, Rahim A, Blum WF, et al.: Hyperleptinaemia in young adults following cranial irradiation in childhood: growth hormone deficiency or leptin insensitivity? Clin Endocrinol (Oxf) 50 (2): 163-9, 1999. [PUBMED Abstract]
Pluimakers VG, van Atteveld JE, de Winter DTC, et al.: Prevalence, risk factors, and optimal way to determine overweight, obesity, and morbid obesity in the first Dutch cohort of 2338 long-term survivors of childhood cancer: a DCCSS-LATER study. Eur J Endocrinol 189 (5): 495-507, 2023. [PUBMED Abstract]
Green DM, Cox CL, Zhu L, et al.: Risk factors for obesity in adult survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. J Clin Oncol 30 (3): 246-55, 2012. [PUBMED Abstract]
Veringa SJ, van Dulmen-den Broeder E, Kaspers GJ, et al.: Blood pressure and body composition in long-term survivors of childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 58 (2): 278-82, 2012. [PUBMED Abstract]
Janiszewski PM, Oeffinger KC, Church TS, et al.: Abdominal obesity, liver fat, and muscle composition in survivors of childhood acute lymphoblastic leukemia. J Clin Endocrinol Metab 92 (10): 3816-21, 2007. [PUBMED Abstract]
Oeffinger KC, Adams-Huet B, Victor RG, et al.: Insulin resistance and risk factors for cardiovascular disease in young adult survivors of childhood acute lymphoblastic leukemia. J Clin Oncol 27 (22): 3698-704, 2009. [PUBMED Abstract]
Jahnukainen K, Heikkinen R, Henriksson M, et al.: Increased Body Adiposity and Serum Leptin Concentrations in Very Long-Term Adult Male Survivors of Childhood Acute Lymphoblastic Leukemia. Horm Res Paediatr 84 (2): 108-15, 2015. [PUBMED Abstract]
Dalton VK, Rue M, Silverman LB, et al.: Height and weight in children treated for acute lymphoblastic leukemia: relationship to CNS treatment. J Clin Oncol 21 (15): 2953-60, 2003. [PUBMED Abstract]
Kohler JA, Moon RJ, Wright S, et al.: Increased adiposity and altered adipocyte function in female survivors of childhood acute lymphoblastic leukaemia treated without cranial radiation. Horm Res Paediatr 75 (6): 433-40, 2011. [PUBMED Abstract]
Zhang FF, Rodday AM, Kelly MJ, et al.: Predictors of being overweight or obese in survivors of pediatric acute lymphoblastic leukemia (ALL). Pediatr Blood Cancer 61 (7): 1263-9, 2014. [PUBMED Abstract]
Warner JT, Evans WD, Webb DK, et al.: Body composition of long-term survivors of acute lymphoblastic leukaemia. Med Pediatr Oncol 38 (3): 165-72, 2002. [PUBMED Abstract]
Miller TL, Lipsitz SR, Lopez-Mitnik G, et al.: Characteristics and determinants of adiposity in pediatric cancer survivors. Cancer Epidemiol Biomarkers Prev 19 (8): 2013-22, 2010. [PUBMED Abstract]
Jarfelt M, Lannering B, Bosaeus I, et al.: Body composition in young adult survivors of childhood acute lymphoblastic leukaemia. Eur J Endocrinol 153 (1): 81-9, 2005. [PUBMED Abstract]
Richard MA, Brown AL, Belmont JW, et al.: Genetic variation in the body mass index of adult survivors of childhood acute lymphoblastic leukemia: A report from the Childhood Cancer Survivor Study and the St. Jude Lifetime Cohort. Cancer 127 (2): 310-318, 2021. [PUBMED Abstract]
Ness KK, DeLany JP, Kaste SC, et al.: Energy balance and fitness in adult survivors of childhood acute lymphoblastic leukemia. Blood 125 (22): 3411-9, 2015. [PUBMED Abstract]
Belle FN, Kasteler R, Schindera C, et al.: No evidence of overweight in long-term survivors of childhood cancer after glucocorticoid treatment. Cancer 124 (17): 3576-3585, 2018. [PUBMED Abstract]
Lindemulder SJ, Stork LC, Bostrom B, et al.: Survivors of standard risk acute lymphoblastic leukemia do not have increased risk for overweight and obesity compared to non-cancer peers: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (6): 1035-41, 2015. [PUBMED Abstract]
Gurney JG, Ness KK, Stovall M, et al.: Final height and body mass index among adult survivors of childhood brain cancer: childhood cancer survivor study. J Clin Endocrinol Metab 88 (10): 4731-9, 2003. [PUBMED Abstract]
Sahakitrungruang T, Klomchan T, Supornsilchai V, et al.: Obesity, metabolic syndrome, and insulin dynamics in children after craniopharyngioma surgery. Eur J Pediatr 170 (6): 763-9, 2011. [PUBMED Abstract]
Müller HL: Childhood craniopharyngioma: current controversies on management in diagnostics, treatment and follow-up. Expert Rev Neurother 10 (4): 515-24, 2010. [PUBMED Abstract]
Simoneau-Roy J, O'Gorman C, Pencharz P, et al.: Insulin sensitivity and secretion in children and adolescents with hypothalamic obesity following treatment for craniopharyngioma. Clin Endocrinol (Oxf) 72 (3): 364-70, 2010. [PUBMED Abstract]
Tosta-Hernandez PDC, Siviero-Miachon AA, da Silva NS, et al.: Childhood Craniopharyngioma: A 22-Year Challenging Follow-Up in a Single Center. Horm Metab Res 50 (9): 675-682, 2018. [PUBMED Abstract]
van Schaik J, van Roessel IMAA, Schouten-van Meeteren NAYN, et al.: High Prevalence of Weight Gain in Childhood Brain Tumor Survivors and Its Association With Hypothalamic-Pituitary Dysfunction. J Clin Oncol 39 (11): 1264-1273, 2021. [PUBMED Abstract]
Neville KA, Cohn RJ, Steinbeck KS, et al.: Hyperinsulinemia, impaired glucose tolerance, and diabetes mellitus in survivors of childhood cancer: prevalence and risk factors. J Clin Endocrinol Metab 91 (11): 4401-7, 2006. [PUBMED Abstract]
Nysom K, Holm K, Michaelsen KF, et al.: Degree of fatness after allogeneic BMT for childhood leukaemia or lymphoma. Bone Marrow Transplant 27 (8): 817-20, 2001. [PUBMED Abstract]
Ketterl TG, Chow EJ, Leisenring WM, et al.: Adipokines, Inflammation, and Adiposity in Hematopoietic Cell Transplantation Survivors. Biol Blood Marrow Transplant 24 (3): 622-626, 2018. [PUBMED Abstract]
Inaba H, Yang J, Kaste SC, et al.: Longitudinal changes in body mass and composition in survivors of childhood hematologic malignancies after allogeneic hematopoietic stem-cell transplantation. J Clin Oncol 30 (32): 3991-7, 2012. [PUBMED Abstract]
Wilson CL, Liu W, Chemaitilly W, et al.: Body Composition, Metabolic Health, and Functional Impairment among Adults Treated for Abdominal and Pelvic Tumors during Childhood. Cancer Epidemiol Biomarkers Prev 29 (9): 1750-1758, 2020. [PUBMED Abstract]
Ness KK, Krull KR, Jones KE, et al.: Physiologic frailty as a sign of accelerated aging among adult survivors of childhood cancer: a report from the St Jude Lifetime cohort study. J Clin Oncol 31 (36): 4496-503, 2013. [PUBMED Abstract]
Hayek S, Gibson TM, Leisenring WM, et al.: Prevalence and Predictors of Frailty in Childhood Cancer Survivors and Siblings: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 38 (3): 232-247, 2020. [PUBMED Abstract]
Delaney A, Howell CR, Krull KR, et al.: Progression of Frailty in Survivors of Childhood Cancer: A St. Jude Lifetime Cohort Report. J Natl Cancer Inst 113 (10): 1415-1421, 2021. [PUBMED Abstract]
Williams AM, Krull KR, Howell CR, et al.: Physiologic Frailty and Neurocognitive Decline Among Young-Adult Childhood Cancer Survivors: A Prospective Study From the St Jude Lifetime Cohort. J Clin Oncol 39 (31): 3485-3495, 2021. [PUBMED Abstract]
van Atteveld JE, de Winter DTC, Pluimakers VG, et al.: Frailty and sarcopenia within the earliest national Dutch childhood cancer survivor cohort (DCCSS-LATER): a cross-sectional study. Lancet Healthy Longev 4 (4): e155-e165, 2023. [PUBMED Abstract]
Guida JL, Hyun G, Belsky DW, et al.: Associations of seven measures of biological age acceleration with frailty and all-cause mortality among adult survivors of childhood cancer in the St. Jude Lifetime Cohort. Nat Cancer 5 (5): 731-741, 2024. [PUBMED Abstract]
Efectos tardíos en el sistema inmunitario
Los efectos tardíos en el sistema inmunitario no se han estudiado bien, en especial en los sobrevivientes sometidos a tratamientos contemporáneos. Los informes publicados sobre los desenlaces a largo plazo en el sistema inmunitario presentan limitaciones debido a la recopilación retrospectiva de datos, el tamaño pequeño de la muestra, el sesgo de selección y participación de la cohorte, la heterogeneidad en el abordaje de tratamiento, el tiempo transcurrido desde el tratamiento y el método de confirmación.
Asplenia
La esplenectomía quirúrgica o funcional aumenta el riesgo de contraer una infección bacteriana invasiva que pone en peligro la vida:[1]
Aunque la laparotomía realizada con propósitos de estadificación ya no es una práctica estándar para el linfoma de Hodgkin infantil, los pacientes de períodos de tratamiento anteriores tienen riesgos que perduran.[2]
Es posible que los niños sometidos a radioterapia dirigida al bazo en dosis mayores de 30 Gy queden asplénicos.[3,4] Las dosis bajas dirigidas al campo de radiación afectado (21 Gy) en combinación con quimioterapia multifarmacológica no parecen influir de manera adversa en el funcionamiento esplénico según las mediciones de las pruebas de glóbulos rojos vacuolados.[4] No se dispone de otros estudios sobre el estado inmunitario después de la administración de radioterapia.
La asplenia funcional (con cuerpos de Howell Jolly, reducción del tamaño del bazo y flujo sanguíneo bajo) después de un trasplante de células madre hematopoyéticas (TCMH) se atribuyó a la enfermedad de injerto contra huésped (EICH).[5]
Los investigadores del Childhood Cancer Survivor Study observaron un aumento significativo del riesgo de mortalidad tardía relacionada con infecciones en sobrevivientes tratados con esplenectomía (riesgo relativo [RR], 7,7; intervalo de confianza [IC] 95 %, 3,1–19,1).[6]
La radiación dirigida al bazo también se relacionó con un riesgo, vinculado a la dosis, de mortalidad tardía por infecciones (0,1–9,9 Gy: RR, 2,0, IC 95 %, 0,9–4,5; 10,0–19,9 Gy: RR, 5,5, IC 95 %, 1,9–15,4; >20,0 Gy: RR, 6,0; IC 95 %, 1,8–20,2).
La incidencia acumulada baja de mortalidad tardía relacionada con infecciones de 1,5 % 35 años después de la esplenectomía y del 0,6 % después de la radiación dirigida al bazo indican que estas situaciones son poco comunes.
En un estudio de cohortes multicéntrico neerlandés participaron sobrevivientes de linfoma de Hodgkin (n = 4919; edad, <51 años en el momento del tratamiento; mediana de seguimiento, 20,2 años) que recibieron tratamiento entre 1965 y 2000.[7]
En el estudio se observó que la mortalidad por enfermedades infecciosas se multiplicó por más de 2, no solo entre los pacientes sometidos a esplenectomía, sino también en los que recibían radioterapia en el bazo.
La mortalidad relacionada con la infección aumentó ligeramente más tras la irradiación del bazo que tras la esplenectomía. Se pensó que esta observación estaba relacionada con la menor tasa de vacunación entre los pacientes que recibieron irradiación del bazo, en comparación con los pacientes a los que se practicó una esplenectomía.
Las personas con asplenia, sin importar la causa del estado asplénico, presentan un aumento del riesgo de bacteriemia fulminante, en especial de bacterias encapsuladas, que se vincula con una tasa alta de mortalidad.[8]
El riesgo de bacteriemia es más alto en los niños pequeños que en los niños más grandes y quizás el riesgo sea más alto durante los años inmediatamente posteriores a una esplenectomía. Sin embargo, la septicemia fulminante se ha notificado en adultos hasta 25 años después de una esplenectomía.
En los niños asplénicos, es posible que la bacteriemia obedezca a los siguientes microorganismos:
Streptococcus pneumoniae. El patógeno más común que causa bacteriemia en niños con asplenia.
Otros estreptococos.
Haemophilus influenzae de tipo b (Hib).
Neisseria meningitidis.
Escherichia coli; Staphylococcus aureus.
Bacilos gramnegativos, como las especies de Salmonella, las especies de Klebsiella y Pseudomonas aeruginosa.
Las personas con asplenia funcional o quirúrgica también tienen un aumento de riesgo de paludismo (malaria) mortal y babesiosis grave.[9]
Atención posterior al tratamiento
Los médicos deben tener en cuenta y fomentar la administración de vacunas inactivadas (por ejemplo, antigripal) y vacunas elaboradas con antígenos purificados (por ejemplo, antineumocócica), componentes bacterianos (por ejemplo, vacuna contra la difteria, el tétanos y la tosferina) o con antígenos recombinados genomanipulados (por ejemplo, antihepatitis B) para todos los sobrevivientes de cáncer y trasplantes, de acuerdo con las dosis y esquemas recomendados.[10-12]
No se ha establecido la eficacia de las vacunas antimeningocócicas para los niños con asplenia.
No hay contraindicaciones conocidas para administrar estas vacunas con jeringas separadas y en sitios diferentes, o al mismo tiempo que otras vacunas obligatorias.
La vacuna antineumocócica conjugada (PCV) y la vacuna antineumocócica polisacárida (PPSV) se indican a la edad recomendada para todos los niños con asplenia.[14,15]
Tras la administración de la cantidad apropiada de dosis de PCV13, se deberá iniciar la administración de PPSV23 a los 24 meses de edad.
Se deberá administrar una segunda dosis 5 años más tarde.
Para niños de 2 a 5 años que recibieron una serie completa de PCV7 y que no recibieron la vacuna PCV13, se debe considerar una dosis suplementaria de PCV13.
Se debe considerar una dosis suplementaria de PCV13, para los niños con asplenia de 6 a 18 años que no hayan recibido una dosis de PCV13.
La vacunación contra Hib se deberá iniciar a los 2 meses de vida, tal como se recomienda para los niños sanos de corta edad y para los niños con asplenia que no se vacunaron antes.[15] Para obtener más información en inglés, consultar Immunization Schedules.[15]
Se recomienda la profilaxis antimicrobiana diaria contra las infecciones neumocócicas en niños pequeños con asplenia, sin importar su estado de vacunación.
Aunque la eficacia de la profilaxis antimicrobiana diaria se ha demostrado solo en pacientes con anemia de células falciformes, esta experiencia se ha extendido a otros niños de riesgo alto, como los niños asplénicos con antecedentes de neoplasias malignas o talasemia.
En general, se deberá considerar la profilaxis antimicrobiana (además de la vacunación) para todos los niños menores de 5 años con asplenia durante por lo menos 1 año después de la esplenectomía.
La edad a la que se suspende la profilaxis antimicrobiana es una decisión empírica.
Sobre la base de un estudio multicéntrico de la enfermedad de células falciformes, la penicilina profiláctica se puede suspender a los 5 años de edad en aquellos que reciben atención médica regular y que no han tenido una infección neumocócica grave o esplenectomía quirúrgica.
No se conoce la duración apropiada de la profilaxis para los niños con asplenia atribuible a otras causas.
Algunos expertos continúan con la profilaxis durante la niñez y la edad adulta; en particular para los pacientes de riesgo alto con asplenia.
En el Cuadro 14 se resumen los efectos tardíos esplénicos y los exámenes médicos de detección correspondientes.
Cuadro 14. Efectos tardíos esplénicosa
Tratamiento predisponente
Efectos inmunitarios
Exámenes médicos de detección e intervenciones
EICH = enfermedad de injerto contra huésped; TCMH = trasplante de células madre hematopoyéticas; IgA = inmunoglobulina A; T = temperatura.
Radioterapia con exposición del bazo, esplenectomía, TCMH con EICH actualmente activa
Asplenia o hiposplenia; sepsis fulminante después de una esplenectomía
Cultivos de sangre durante episodios febriles (T >38,5°C), antibióticos empíricos
Vacunación contra organismos encapsulados (vacunas antineumocócicas, contra Haemophilus influenzae tipo b y antimeningocócicas).
TCMH con cualquier antecedente de EICH crónica
Complicaciones inmunitarias (deficiencias de la IgA secretora, hipogammaglobulinemia, disminución de células B, disfunción de células T, infecciones crónicas [por ejemplo, conjuntivitis, sinusitis y bronquitis relacionadas con EICH crónica])
Antecedentes: conjuntivitis crónica, sinusitis crónica, bronquitis crónica, infecciones recidivantes o no habituales, y sepsis
Hallazgos del examen: revisión minuciosa de los ojos, la nariz, los senos paranasales y los pulmones
Aunque el sistema inmunitario se recupera de los efectos de la quimioterapia y la radioterapia activas, su efecto sobre la función inmunitaria en la supervivencia no se ha estudiado bien. En la mayoría de los estudios se ha evaluado a niños tratados por leucemia linfoblástica aguda (LLA) que, en la propia enfermedad, implica una alteración de la maduración linfocítica de las células T o B, además de la terapia linfotóxica.[16] En estudios pequeños se indica que los valores de protección frente a enfermedades prevenibles mediante vacunación pueden verse afectados durante meses o años tras el tratamiento del cáncer infantil sin TCMH. No existen normas universales sobre el calendario de las evaluaciones serológicas ni sobre si deben administrarse refuerzos y revacunaciones a todos los sobrevivientes.[16]
Se observaron defectos en la recuperación inmunitaria caracterizados por la disminución del número de células B al cabo de 2 años en sobrevivientes de LLA de riesgo estándar e intermedio.[17]
En un estudio retrospectivo de 55 sobrevivientes de cáncer infantil que recibieron quimioterapia estándar, más del 75 % de los sobrevivientes a largo plazo que recibieron al menos una vacuna contra el sarampión, la parotiditis y la rubéola (SPR) antes del diagnóstico de cáncer carecían de seroprotección frente a los virus del sarampión y la parotiditis. Esto indica que el calendario interrumpido no es suficiente para la seroprotección a largo plazo. Todos los sobrevivientes de cáncer infantil que recibieron las 2 dosis de la vacuna triple vírica una vez finalizado el tratamiento contra el cáncer presentaron valores positivos frente a los virus del sarampión y la parotiditis.[18]
Los sobrevivientes de cáncer infantil a veces permanecen susceptibles a infecciones prevenibles mediante vacunas. La intensidad del tratamiento, la edad en el momento del diagnóstico y el tiempo transcurrido desde el tratamiento se relacionan con el riesgo de perder la inmunidad previa.[19,20]
Si bien hay una escasez de datos sobre los beneficios de la administración de vacunas activas en esta población, la revacunación es necesaria para proporcionar anticuerpos protectores. El calendario de revacunación recomendado dependerá de las vacunas recibidas previamente y de la intensidad de la terapia.[16]
En algunos niños que recibieron tratamiento intensivo, tal vez sea aconsejable evaluar los anticuerpos contra los antígenos de vacunación comunes para determinar la necesidad de revacunación. Para obtener más información en inglés, consultar Immunization Schedules.[15]
El estado inmunitario también se ve comprometido después de un TCMH, en particular cuando se relaciona con la EICH.[21]
En un estudio prospectivo longitudinal de 210 sobrevivientes tratados con TCMH alogénico, se observaron respuestas de anticuerpos que se prolongaron durante más de 5 años después de la vacunación contra tétanos (95,7 %), rubéola (92,3 %), poliovirus (97,9 %) y en quienes recibieron la vacuna acelular contra la difteria, el tétanos y la tosferina (DTaP) (100 %).[22]
Las respuestas a la tosferina (25,0 %), el sarampión (66,7 %), la parotiditis infecciosa (61,5 %), la hepatitis B (72,9 %) y la difteria (48,6 %) en quienes recibieron la vacuna contra el tétanos y la difteria (Td) fueron menos favorables.
Los factores relacionados con el fracaso de la vacunación incluyen edad mayor en el momento de la vacunación, recuento más bajo de CD3, CD4 o CD19; concentración más alta de inmunoglobulina (lg) M; serología positiva para el citomegalovirus en los receptores; valores de anticuerpos negativos antes de la vacunación; antecedentes de EICH aguda o crónica y acondicionamiento con radiación.
Los principales grupos de trasplante de América del Norte y Europa, los CDC y la Infectious Diseases Society of America publicaron recomendaciones para el seguimiento de los receptores de trasplantes.[23-25] La American Society for Transplantation and Cellular Therapy ha emitido recomendaciones para prevenir el sarampión en pacientes que recibieron TCMH o terapia de células T con receptores de antígenos quiméricos (CAR).[26]
Se desconocen en gran medida los efectos tardíos tras el tratamiento con células T CAR dirigidas a CD19 en pacientes con LLA recidivante o resistente al tratamiento. Muchos pacientes se someterán a un TCMH después del tratamiento con células CAR-T, lo que complica la evaluación de los efectos tardíos. El efecto tardío más frecuente (definido a los 90 días tras la terapia con células T CAR) es la hipogammaglobulinemia.
En una serie de 75 niños que recibieron terapia con células T CAR, todos los pacientes que respondieron al tratamiento (n = 61) experimentaron aplasia de células B. Se observó un 83 % de probabilidad de aplasia de células B en curso a los 6 meses del tratamiento con tisagenlecleucel. El 90 % de los pacientes que respondieron al tratamiento con células T CAR necesitaron IgG intravenosa (IV).[27]
En una serie se incluyeron 86 pacientes adultos (se incluyeron adultos jóvenes de 22 años o más) tratados con células T CAR para enfermedades resistentes al tratamiento o recidivantes positivas para CD19 (LLA, leucemia linfocítica crónica y linfoma no Hodgkin) que permanecieron vivos durante al menos 1 año tras el tratamiento. Se observaron efectos tardíos que comenzaron o persistieron más de 90 días después del tratamiento con células T CAR. Con los datos disponibles, se observó hipogammaglobulinemia (IgG <400 mg/dl o sustitución de gammaglobulina IV) en el 67 % de los pacientes.[28]
La inmunidad humoral preexistente frente a antígenos relacionados con vacunas puede persistir en pacientes a pesar de la marcada aplasia de células B tras la inmunoterapia con células T dirigidas a CD19. Los estudios han demostrado que una población de células plasmáticas que carecen de expresión de CD19 sobrevive a largo plazo tras la inmunoterapia con células T CAR CD19.[29]
Riesgos de infección
Las complicaciones infecciosas que dan lugar a la hospitalización se producen en tasas excesivas entre los sobrevivientes de cáncer infantil a largo plazo.[30]
En un estudio retrospectivo de observación poblacional, la tasa de incidencia fue de 12,6 por 1000 años-persona para la infección relacionada con la hospitalización en sobrevivientes a 5 años de cáncer infantil, en comparación con 2,4 para las personas sin cáncer que se emparejaron por año de nacimiento y sexo. Esto representó una razón de tasas de incidencia de 5,1.
Las tasas de incidentes se mantuvieron elevadas más del doble durante los períodos de 5 a 10 años y más de 10 años después del diagnóstico.
Los sobrevivientes tuvieron un riesgo más alto de infección (razón de tasas de incidencia, 13,1), en comparación con los comparadores cuando se evaluaron infecciones potencialmente prevenibles por vacunación.
Centers for Disease Control and Prevention: Altered immunocompetence. In: Kroger A, Bahta L, Long S, et al.: General Best Practice Guidelines for Immunization: Best Practices Guidance. Atlanta, GA: Advisory Committee on Immunization Practices (ACIP); Centers for Disease Control and Prevention, 2023, pp 123-48. Available online. Last accessed November 3, 2023.
Jockovich M, Mendenhall NP, Sombeck MD, et al.: Long-term complications of laparotomy in Hodgkin's disease. Ann Surg 219 (6): 615-21; discussion 621-4, 1994. [PUBMED Abstract]
Coleman CN, McDougall IR, Dailey MO, et al.: Functional hyposplenia after splenic irradiation for Hodgkin's disease. Ann Intern Med 96 (1): 44-7, 1982. [PUBMED Abstract]
Weiner MA, Landmann RG, DeParedes L, et al.: Vesiculated erythrocytes as a determination of splenic reticuloendothelial function in pediatric patients with Hodgkin's disease. J Pediatr Hematol Oncol 17 (4): 338-41, 1995. [PUBMED Abstract]
Al-Eid MA, Tutschka PJ, Wagner HN, et al.: Functional asplenia in patients with chronic graft-versus-host disease: concise communication. J Nucl Med 24 (12): 1123-6, 1983. [PUBMED Abstract]
Weil BR, Madenci AL, Liu Q, et al.: Late Infection-Related Mortality in Asplenic Survivors of Childhood Cancer: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 36 (16): 1571-1578, 2018. [PUBMED Abstract]
de Vries S, Schaapveld M, Janus CPM, et al.: Long-Term Cause-Specific Mortality in Hodgkin Lymphoma Patients. J Natl Cancer Inst 113 (6): 760-769, 2021. [PUBMED Abstract]
Madenci AL, Armstrong LB, Kwon NK, et al.: Incidence and risk factors for sepsis after childhood splenectomy. J Pediatr Surg 54 (7): 1445-1448, 2019. [PUBMED Abstract]
Ram S, Lewis LA, Rice PA: Infections of people with complement deficiencies and patients who have undergone splenectomy. Clin Microbiol Rev 23 (4): 740-80, 2010. [PUBMED Abstract]
National Center for Immunization and Respiratory Diseases: General recommendations on immunization: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 60 (RR02): 1-60, 2011. Available online. Last accessed August 21, 2023.
Bridges CB, Coyne-Beasley T; Advisory Committee on Immunization Practices: Advisory committee on immunization practices recommended immunization schedule for adults aged 19 years or older: United States, 2014. Ann Intern Med 160 (3): 190, 2014. [PUBMED Abstract]
Rubin LG, Levin MJ, Ljungman P, et al.: 2013 IDSA clinical practice guideline for vaccination of the immunocompromised host. Clin Infect Dis 58 (3): 309-18, 2014. [PUBMED Abstract]
Mbaeyi SA, Bozio CH, Duffy J, et al.: Meningococcal Vaccination: Recommendations of the Advisory Committee on Immunization Practices, United States, 2020. MMWR Morb Mortal Wkly Rep 69 (No. RR-9): 1–41, 2020. Available online. Last accessed November 3, 2023.
Centers for Disease Control and Prevention (CDC): Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine among children aged 6-18 years with immunocompromising conditions: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 62 (25): 521-4, 2013. [PUBMED Abstract]
Centers for Disease Control and Prevention: Child and Adolescent Immunization Schedule by Medical Indication: Recommendations for Ages 18 Years or Younger, United States, 2025. Atlanta, GA: Centers for Disease Control and Prevention, 2025. Available online. Last accessed January 6, 2025.
Guilcher GMT, Rivard L, Huang JT, et al.: Immune function in childhood cancer survivors: a Children's Oncology Group review. Lancet Child Adolesc Health 5 (4): 284-294, 2021. [PUBMED Abstract]
Koskenvuo M, Ekman I, Saha E, et al.: Immunological Reconstitution in Children After Completing Conventional Chemotherapy of Acute Lymphoblastic Leukemia is Marked by Impaired B-cell Compartment. Pediatr Blood Cancer 63 (9): 1653-6, 2016. [PUBMED Abstract]
Speckhart SA: MMR vaccination timing and long-term immunity among childhood cancer survivors. Pediatr Blood Cancer 70 (4): e30133, 2023. [PUBMED Abstract]
Fayea NY, Fouda AE, Kandil SM: Immunization status in childhood cancer survivors: A hidden risk which could be prevented. Pediatr Neonatol 58 (6): 541-545, 2017. [PUBMED Abstract]
Bochennek K, Allwinn R, Langer R, et al.: Differential loss of humoral immunity against measles, mumps, rubella and varicella-zoster virus in children treated for cancer. Vaccine 32 (27): 3357-61, 2014. [PUBMED Abstract]
Olkinuora HA, Taskinen MH, Saarinen-Pihkala UM, et al.: Multiple viral infections post-hematopoietic stem cell transplantation are linked to the appearance of chronic GVHD among pediatric recipients of allogeneic grafts. Pediatr Transplant 14 (2): 242-8, 2010. [PUBMED Abstract]
Inaba H, Hartford CM, Pei D, et al.: Longitudinal analysis of antibody response to immunization in paediatric survivors after allogeneic haematopoietic stem cell transplantation. Br J Haematol 156 (1): 109-17, 2012. [PUBMED Abstract]
Tomblyn M, Chiller T, Einsele H, et al.: Guidelines for preventing infectious complications among hematopoietic cell transplantation recipients: a global perspective. Biol Blood Marrow Transplant 15 (10): 1143-238, 2009. [PUBMED Abstract]
Cordonnier C, Einarsdottir S, Cesaro S, et al.: Vaccination of haemopoietic stem cell transplant recipients: guidelines of the 2017 European Conference on Infections in Leukaemia (ECIL 7). Lancet Infect Dis 19 (6): e200-e212, 2019. [PUBMED Abstract]
Majhail NS, Rizzo JD, Lee SJ, et al.: Recommended screening and preventive practices for long-term survivors after hematopoietic cell transplantation. Rev Bras Hematol Hemoter 34 (2): 109-33, 2012. [PUBMED Abstract]
Pergam SA, Englund JA, Kamboj M, et al.: Preventing Measles in Immunosuppressed Cancer and Hematopoietic Cell Transplantation Patients: A Position Statement by the American Society for Transplantation and Cellular Therapy. Biol Blood Marrow Transplant 25 (11): e321-e330, 2019. [PUBMED Abstract]
Maude SL, Laetsch TW, Buechner J, et al.: Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 378 (5): 439-448, 2018. [PUBMED Abstract]
Cordeiro A, Bezerra ED, Hirayama AV, et al.: Late Events after Treatment with CD19-Targeted Chimeric Antigen Receptor Modified T Cells. Biol Blood Marrow Transplant 26 (1): 26-33, 2020. [PUBMED Abstract]
Bhoj VG, Arhontoulis D, Wertheim G, et al.: Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood 128 (3): 360-70, 2016. [PUBMED Abstract]
Chehab L, Doody DR, Esbenshade AJ, et al.: A Population-Based Study of the Long-Term Risk of Infections Associated With Hospitalization in Childhood Cancer Survivors. J Clin Oncol 41 (2): 364-372, 2023. [PUBMED Abstract]
Efectos tardíos en el sistema osteomuscular
El sistema osteomuscular de niños y adolescentes en crecimiento es vulnerable a los efectos citotóxicos de los tratamientos del cáncer, como cirugía, quimioterapia y radioterapia. Los efectos tardíos documentados son los siguientes:
Problemas en los huesos y las articulaciones (crecimiento anormal de los huesos o los músculos).
Deformidades y pérdidas funcionales relacionadas con la cirugía de amputación o conservación del miembro, contractura articular, osteoporosis o fracturas y osteonecrosis.
Cambios en la composición corporal (obesidad y pérdida de masa muscular magra).
Si bien estos efectos tardíos se describen por separado, es importante tener en cuenta que todos los componentes del sistema osteomuscular se interrelacionan. Por ejemplo, la hipoplasia de un grupo muscular perjudica el funcionamiento de los huesos largos y la disfunción resultante tal vez produzca después inactividad y osteoporosis.
La fortaleza principal de la bibliografía publicada en la que se documentan efectos tardíos osteomusculares en los niños y adolescentes tratados por cáncer es que la mayoría de los estudios presentan desenlaces y exposiciones claramente definidos. Sin embargo, muchos de los diseños de los estudios son observacionales y transversales, o retrospectivos. Los estudios de una sola institución son comunes y, para algunos desenlaces, solo se describieron pequeñas cohortes de conveniencia. De este modo, es posible que se excluyera de los estudios a pacientes con los efectos osteomusculares más graves por defunción o imposibilidad de participar en las pruebas de seguimiento, o que se presentaran muestras excesivas de aquellos con los efectos tardíos osteomusculares más graves dado que estos pacientes estuvieron disponibles al regresar para el seguimiento de complicaciones relacionadas con el tratamiento. Además, algunos de los desenlaces notificados en adultos sobrevivientes de cáncer infantil quizá no sean importantes para los pacientes que están en tratamiento en la actualidad en vista de los cambios en el tratamiento contra el cáncer y la administración de modalidades de tratamientos contra el cáncer en respuesta a los efectos tóxicos documentados; en particular, la radioterapia.
Crecimiento óseo anormal
El efecto de la radiación en el crecimiento óseo depende de los sitios irradiados, como los siguientes:
La radiación puede inhibir la maduración y el desarrollo normales de los huesos y los músculos, de una manera que depende de la edad y de la dosis.
La radiación dirigida a la cabeza (por ejemplo, radioterapia craneal, orbital, infratemporal o nasofaríngea) puede causar anomalías craneofaciales, en particular, en niños tratados antes de los 5 años o con dosis de radiación de 20 Gy o más,.[1-4] o que se trataron con quimioterapia simultánea.[5]
Los sarcomas de tejido blando, como el rabdomiosarcoma orbitario y el retinoblastoma, son dos de los tipos más comunes de cánceres tratados con estos campos de radiación. A menudo, además del efecto cosmético de las anomalías craneofaciales, se presentan problemas relacionados con la dentadura y los senos paranasales.
La radioterapia craneal daña el sistema hipotalamohipofisario de una manera que se relaciona con la edad y la respuesta a la dosis, y es posible que produzca deficiencia de la hormona del crecimiento.[6,7] Si no se trata esta deficiencia durante los años de crecimiento, o algunas veces incluso cuando se administra el tratamiento apropiado, la radioterapia craneal conduce a una estatura final mucho más baja. Los pacientes con un tumor en el sistema nervioso central (SNC) [8,9] o leucemia linfoblástica aguda (LLA) [10,11] tratados con 18 Gy o más de radioterapia craneal tienen el riesgo más alto. Los pacientes tratados con irradiación corporal total (ICT) de fracción única,[12-14] y aquellos tratados con radiación craneal para tumores sólidos fuera del SNC [15] también tienen riesgo de deficiencia de la hormona del crecimiento. Si además se irradia la columna vertebral (por ejemplo, radioterapia craneoespinal por meduloblastoma o tratamientos anteriores administrados en la década de 1960 para la LLA), el crecimiento se ve afectado por dos mecanismos separados: deficiencia de la hormona del crecimiento y daño directo a la columna vertebral.
Radiación dirigida a la columna vertebral y los huesos largos
También es posible que la radioterapia afecte de forma directa el crecimiento de la columna vertebral y los huesos largos (así como los grupos musculares relacionados con ellos), y que a veces provoque el cierre prematuro de las epífisis, lo que da lugar a las siguientes características:[16-18]
Estatura baja.
Crecimiento asimétrico (escoliosis o cifosis).
Discrepancias en la longitud de las extremidades.
La radioterapia con ortovoltaje, que se usaba de manera frecuente antes de 1970, enviaba dosis altas de radiación a los huesos y era habitual que se relacionara con anomalías subsiguientes del crecimiento óseo.
Sin embargo, incluso con la radioterapia contemporánea, si un tumor sólido se localiza cerca de una epífisis o la columna vertebral, tal vez sea difícil evitar alteraciones en el desarrollo óseo.
Se evaluaron los efectos de la radioterapia dirigida a la columna vertebral en sobrevivientes de tumores del SNC y Wilms.
Evidencia (efecto de la radioterapia en la columna vertebral y los huesos largos):
Se evaluó el efecto de la irradiación craneoespinal (ICS) sobre el crecimiento óseo en 220 pacientes pediátricos con tumores cerebrales que recibieron tratamiento con ICS.[19]
El crecimiento vertebral se correlacionó de manera significativa con la dosis de radiación (>39 Gy) y la deficiencia de hormona del crecimiento.
De los pacientes, el 83 % presentó cambios en la médula grasa, el 31 % cambios degenerativos en los discos (cambio de señal, abultamiento y pérdida de altura), el 13 % cambios degenerativos de los huesos de la columna, el 17 % acuñamiento o pérdida de altura vertebral y el 27 % escoliosis.
Incluso cuando se administró irradiación simétrica dirigida a los cuerpos vertebrales, el riesgo de escoliosis fue más alto después de la ICE.
La edad en el momento de la irradiación fue un factor de riesgo significativo de escoliosis (P = 0,0067), pero no se correlacionó con la dosis de radiación.
El reemplazo adecuado de la hormona del crecimiento fue importante para el crecimiento de los huesos.
En el National Wilms Tumor Study (estudios del 1 al 4), se evaluó la pérdida de estatura en 2778 niños.[20]
Para aquellos menores de 12 meses en el momento del diagnóstico que recibieron más de 10 Gy, la deficiencia calculada de estatura adulta fue de 7,7 cm cuando se lo comparó con el grupo que no recibió radioterapia.
Para quienes recibieron 10 Gy, se calculó que el acortamiento del tronco fue de 2,8 cm o menos.
Entre aquellos cuyas mediciones de estatura durante la adolescencia estaban disponibles, los pacientes que recibieron más de 15 Gy de radioterapia fueron, en promedio, de 4 cm a 7 cm más bajos que sus contrapartes no irradiados, con una clara relación entre la dosis y la respuesta.
La quimioterapia no confirió un riesgo adicional.
También se volvió a evaluar el efecto de la radioterapia en la presentación de escoliosis. En un grupo de 42 niños tratados por tumor de Wilms desde 1968 a 1994, se observó escoliosis en 18 pacientes; solo un paciente necesitó una intervención ortopédica. En el estudio también se indicó que la incidencia fue menor en pacientes que recibieron entre 10 y 12 Gy, que son las dosis que se usan de manera habitual para el tumor de Wilms. Sin embargo, el tamaño de la muestra era pequeño.[21]
En una cohorte de 245 sobrevivientes de LLA a largo plazo (mediana de tiempo de seguimiento, 15,1 años), se evaluó la prevalencia y los factores de riesgo de las deformidades vertebrales mediante radiografías de la columna vertebral y exploraciones con absorciometría dual de rayos X.[22]
Las deformidades vertebrales eran prevalentes en el 23 % de los pacientes, y el 88 % de ellas se clasificaron como de grado 1 de Genant (>reducción del 20–25 % en el porcentaje de las relaciones de altura vertebral anterior, media o posterior).
Los predictores independientes de deformidad vertebral prevalente incluían el sexo masculino (riesgo relativo [RR], 1,94; IC 95 %, 1,16–3,24), una mayor dosis acumulada de glucocorticoides (RR, 1,05; IC 95 %, 1,00–1,10) y el dolor de espalda (RR, 2,44; IC 95 %, 1,56–3,84).
La puntuación Z de la densidad mineral ósea de la columna lumbar y la edad y el tiempo transcurridos desde el diagnóstico no fueron factores predictivos significativos de la deformidad vertebral. Sin embargo, los análisis estratificados por sexo indicaron que la puntuación Z de la columna lumbar predecía significativamente la deformidad vertebral en los hombres.
Osteoporosis y fracturas
Aunque no se ha notificado un aumento de las tasas de fracturas de los sobrevivientes a largo plazo de cáncer infantil,[23] la masa ósea máxima es un factor importante que influye en el riesgo de osteoporosis y fracturas en pacientes de edad más avanzada. Los factores relacionados con el tratamiento que afectan la pérdida mineral ósea son los siguientes:
Quimioterapia. El metotrexato tiene un efecto citotóxico en los osteoblastos; esto produce una reducción del volumen óseo y la formación de hueso nuevo.[24] Es posible que este efecto se exacerbe por el uso crónico de corticoesteroides, otra clase de fármacos que se usan de modo rutinario para tratar neoplasias hematológicas malignas y para los tratamientos de apoyo de una variedad de cánceres infantiles.
Radioterapia. Las endocrinopatías relacionadas con la radiación, como la deficiencia de la hormona del crecimiento o el hipogonadismo, quizás contribuyan a una pérdida constante de minerales óseos.[25,26]
Nutrición y actividad. La nutrición subóptima y la inactividad física predisponen aún más a deficiencias en la acumulación de minerales óseos.
La mayor parte del conocimiento disponible acerca de los efectos del cáncer y su tratamiento sobre la mineralización ósea se derivó de estudios de niños con LLA.
En niños con LLA el proceso leucémico y la deficiencia de vitamina D quizás cumplan una función en las alteraciones del metabolismo óseo y de la masa ósea observadas en el momento del diagnóstico.[27]
El tratamiento antileucémico produce pérdida adicional de la densidad mineral ósea;[28] esto se normaliza con el paso del tiempo [29] o persiste durante muchos años después de finalizar el tratamiento.[30]
Los factores clínicos de predicción de un riesgo más alto de baja densidad mineral ósea comprenden los siguientes tratamientos:[31-33]
Dosis acumuladas altas de metotrexato (>40 g/m2).
Dosis acumuladas altas de corticoesteroides (>9 g/m2).
Radioterapia craneal o craneoespinal.
Glucocorticoides más potentes, como la dexametasona.
La presentación de osteonecrosis durante el tratamiento de la LLA.[34]
La evaluación clínica de la densidad mineral ósea en adultos tratados por LLA infantil indica que la mayoría de las deficiencias minerales óseas se normalizan con el tiempo tras la interrupción del tratamiento osteotóxico.[32]
Evidencia (densidad mineral ósea baja):
Los investigadores del Dutch Childhood Oncology Group (DCOG)–LATER evaluaron los factores de riesgo de densidad mineral ósea baja y muy baja, cualquier fractura y fractura vertebral con absorciometría dual de rayos X en 3996 sobrevivientes de cáncer infantil a 5 años (media de edad, 33 años en el momento de la participación). Se evaluaron las fracturas autonotificadas en todos los participantes y las fracturas vertebrales en un subgrupo de participantes.[26]
La razón de incidencia estandarizada de cualquier primera fractura fue de 3,53 (intervalo de confianza [IC] 95 %, 3,06–4,06) para los hombres participantes y de 5,35 (4,46–6,52) para las mujeres que participaron. De 249 participantes, 33 (13,3 %) presentaron fracturas vertebrales.
Los factores de riesgo de densidad mineral ósea baja en cualquier sitio fueron el sexo masculino, el peso insuficiente, la exposición a dosis altas de carboplatino, la exposición a cualquier dosis de radioterapia craneal, el hipogonadismo, el hipertiroidismo, la actividad física baja y la deficiencia grave de vitamina D.
Los factores de riesgo de densidad muy baja de la médula ósea en cualquier sitio fueron el sexo masculino, el peso insuficiente, la exposición a radioterapia craneal, la deficiencia de la hormona del crecimiento y la deficiencia grave de vitamina D.
Los factores de riesgo de cualquier fractura fueron el sexo masculino, la situación de consumo de tabaco anterior y actual, y la densidad mineral ósea de la columna lumbar muy baja.
Los factores de riesgo de fracturas vertebrales fueron la edad más avanzada en el momento del seguimiento, el tratamiento previo con compuestos derivados del platino, la deficiencia de la hormona del crecimiento y la actividad física baja.
Se evaluó el déficit de densidad mineral ósea en 3919 sobrevivientes a 5 años (mediana de edad, 31,7 años) en la cohorte del St. Jude Life (SJLIFE). La exposición al tratamiento, las afecciones concomitantes y el estado de consumo de tabaco o sedentarismo explicaron el 18,5 %, el 10,2 % y el 7,0 % de los casos moderados de déficit de densidad mineral ósea, respectivamente. Estos elementos también explicaron el 55,4 %, el 51,1 % y el 9,9 % de los casos graves de déficit de densidad mineral ósea, respectivamente.[35]
Los déficits graves de densidad mineral ósea se relacionaron con la radioterapia dirigida al cráneo de 30 Gy o superior (OR, 5,22), radioterapia dirigida a los testículos o a la pelvis (OR, 1,70), hipogonadismo (OR, 3,27), deficiencia de la hormona de crecimiento (OR, 5,28), consumo de tabaco (OR, 1,70) y sedentarismo (OR, 2,06).
Los sobrevivientes con déficit de densidad mineral ósea fueron menos propensos a vivir solos o tener un trabajo. Además, fue más probable que necesitaran ayuda con el cuidado personal, notificaran síntomas de depresión y comunicaran una calidad de vida precaria.
Se notificaron reducciones de densidad mineral ósea en sobrevivientes a largo plazo de tumores de encéfalo infantiles.[36]
Entre 50 sobrevivientes de meduloblastoma o tumores neuroectodérmicos primitivos supratentoriales del SNC (mediana de seguimiento, 19,5 años), se detectaron puntuaciones z de densidad de médula ósea disminuidas en un 48 % de la columna lumbar y en un 34 % de las mediciones corporales totales.
Los factores de riesgo modificables de la reducción de densidad de la médula ósea fueron concentraciones bajas de calcio sérico (62 %) y vitamina D (26 %), el consumo bajo de vitamina D (69 %) y el estilo de vida inactivo (62 %).
El sexo masculino, la edad más avanzada en el momento del diagnóstico y un índice de masa muscular (IMC) magro más bajo se relacionaron con un aumento del riesgo de disminución de los puntuaciones z de la densidad de la médula ósea.
Entre 862 sobrevivientes de LLA (mediana de edad, 31,3 años) evaluados mediante tomografía computarizada cuantitativa en las vértebras L1 hasta L2, se notificó lo siguiente:[25]
El 30 % de los sobrevivientes presentaron densidad mineral ósea baja (puntuación z inferior a -1), y el 18,6 % de los sobrevivientes cumplieron con los criterios de fragilidad o prefragilidad.
El fenotipo de prefragilidad se caracteriza por tener 2 de 5 características (masa muscular baja, agotamiento autonotificado, gasto energético bajo, velocidad de marcha lenta y debilidad) y el fenotipo frágil se caracteriza por tener 3 o más de estas características.
Los factores modificables como la deficiencia de la hormona del crecimiento, el consumo de tabaco y el consumo de bebidas alcohólicas fueron factores pronósticos significativos de estos desenlaces, con un efecto variable según el sexo.
Estos datos subrayan la importancia de la orientación sobre el estilo de vida y los exámenes de detección de las deficiencias hormonales durante las evaluaciones de seguimiento de los sobrevivientes a largo plazo.
Una cohorte de 845 adultos sobrevivientes de la LLA infantil se evaluaron a una mediana de edad de 31 años.[32]
La densidad mineral ósea muy baja fue relativamente poco frecuente; solo el 5,7 % de los pacientes exhibieron puntuaciones z de densidad mineral ósea compatibles con osteoporosis y en el 23,8 %, con osteopenia.
La dosis de radiación craneal de 24 Gy o más, pero no las dosis acumuladas de metotrexato o equivalentes de prednisona, se relacionó con la duplicación del riesgo de puntuaciones z de densidad mineral ósea de -1 o inferiores.
En un subconjunto de 400 sobrevivientes con evaluaciones de densidad mineral ósea longitudinal, las puntuaciones z de la densidad mineral ósea tendieron a mejorar desde la adolescencia a la edad adulta joven.
Se ha notificado que los receptores de trasplantes de células hematopoyéticas (TCMH) alogénicos condicionados con ICT presentan deficiencias en la densidad mineral ósea, que es probable que tengan causas multifactoriales.
Investigadores franceses observaron un riesgo significativo de densidad mineral ósea femoral más baja entre adultos sobrevivientes de leucemia infantil tratados con TCMH que tenían deficiencia gonadal.[37]
Se observó que la terapia hormonal mejora la densidad mineral ósea de las adolescentes con diagnóstico de hipogonadismo después de un TCMH.[38]
En un estudio transversal se evaluó la prevalencia y los factores de riesgo de la deficiencia de vitamina D (<20 ng/ml), así como su asociación con la densidad mineral ósea en 446 sobrevivientes de cáncer infantil a largo plazo (mediana de edad, 27,5 años; mediana de tiempo desde completar el tratamiento, 14,2 años).[39]
Se presentó deficiencia de vitamina D en el 24 % de los sobrevivientes.
Los factores de predicción independientes de la deficiencia de vitamina D fueron un IMC alto (OR, 1,78 en pacientes con sobrepeso; OR, 2,4 en pacientes con obesidad) y la raza y etnia hispana o negra (OR, 2,4).
El riesgo de disminución de la densidad mineral ósea fue 3 veces más alto (OR, 3,58) en los sobrevivientes con deficiencia de vitamina D.
Riesgo de fracturas en sobrevivientes de cáncer infantil
A pesar de los riesgos relacionados con la enfermedad y el tratamiento de las deficiencias de densidad mineral ósea, la prevalencia de las fracturas autonotificadas de los participantes del Childhood Cancer Survivor Study (CCSS) fue más baja que la notificada por los hermanos del grupo de control. Los factores de predicción de aumento de prevalencia de fracturas identificados mediante análisis multivariantes fueron los siguientes:[23]
En las mujeres sobrevivientes, mayor edad en el momento del seguimiento, raza blanca, tratamiento con metotrexato y alteraciones del equilibrio.
En los varones sobrevivientes, antecedentes de consumo de tabaco y raza blanca.
Las fracturas inducidas por radiación se presentan con dosis de radiación de 50 Gy o superiores, que se usan a menudo para tratar el sarcoma de Ewing en las extremidades.[40]
Modelo de predicción del riesgo de deficiencia de densidad mineral ósea
Se utilizaron los datos del estudio de cohorte del SJLIFE (formulación) y del Erasmus Medical Center (validación) en los Países Bajos para formular y validar modelos de predicción para la densidad mineral ósea baja o muy baja según las características clínicas y terapéuticas que identifican a los adultos sobrevivientes de cáncer infantil que requieren detección mediante absorciometría de rayos X de doble energía.[41]
La densidad mineral ósea baja se definió como puntuación z de -1 o menos para la densidad mineral ósea de la columna lumbar o de todo el cuerpo; la densidad mineral ósea muy baja se definió como una puntuación z de -2 o menos.
Se detectó densidad mineral ósea baja en el 51 % de los participantes del estudio SJLIFE y en el 45 % de los participantes neerlandeses, lo que abarcó un grupo de sobrevivientes de neoplasias malignas hematológicas y sólidas. Se detectó densidad mineral ósea muy baja en el 20 % de los participantes del estudio SJLIFE y en el 10 % de los participantes neerlandeses.
En el modelo, que incluyó sexo masculino, estatura, peso, edad, situación actual de consumo de tabaco e irradiación craneal, se demostraron buenos resultados para predecir el riesgo de densidad mineral ósea baja (áreas bajo la curva de 0,72 en la cohorte SJLIFE y de 0,69 en la cohorte neerlandesa).
En el modelo, que incluyó sexo masculino, estatura, peso, edad, irradiación craneal e irradiación abdominal, se demostraron buenos resultados para predecir el riesgo de densidad mineral ósea muy baja (áreas bajo la curva de 0,76 en la cohorte SJLIFE y de 0,75 en la cohorte neerlandesa).
Mediante estos modelos se identificaron de forma correcta los estados de densidad mineral ósea en la mayoría de los adultos sobrevivientes blancos de hasta 40 años utilizando las características del paciente y del tratamiento, medidas con facilidad.
La calculadora para determinar la probabilidad prevista de densidad mineral ósea baja y muy baja para un sobreviviente individual está disponible en inglés en Internet.
Osteonecrosis
La osteonecrosis (conocida también como necrosis aséptica o avascular) es una complicación esquelética poco frecuente, pero bien reconocida, que se observa sobre todo en sobrevivientes de neoplasias malignas hematológicas tratados con corticoesteroides.[42] La afección se caracteriza por la muerte de uno o más segmentos de hueso que con mayor frecuencia afectan las articulaciones que soportan peso, en especial las caderas y las rodillas.
La prevalencia de osteonecrosis osciló entre el 1 % y el 22 % según la población estudiada, el protocolo de tratamiento, el método de evaluación y el tiempo transcurrido desde el tratamiento.[42-48]
En estudios longitudinales de cohortes, se identificó una gama de manifestaciones clínicas de osteonecrosis, que oscilan desde cambios asintomáticos y de resolución espontánea en las imágenes hasta un colapso articular progresivo doloroso que exige el reemplazo articular.[49]
La osteonecrosis sintomática, caracterizada por dolor, inflamación articular y reducción de la movilidad, por lo común se presenta durante los primeros 2 años de tratamiento; en particular, en pacientes de LLA.
Estos síntomas tal vez mejoren con el tiempo, persistan o progresen en los años posteriores a la finalización del tratamiento.[50] En una serie, el 60 % de los pacientes continuaron sintomáticos al cabo de una mediana de seguimiento de 4,9 años después del diagnóstico de la osteonecrosis.[51]
Se ha observado el deterioro del rendimiento físico y la reducción de la calidad de vida en sobrevivientes a largo plazo de leucemia y linfoma infantil (media de edad, 28 años) con osteonecrosis.[52]
En los pacientes en los que persisten los síntomas graves, algunas veces se realizan procedimientos quirúrgicos, como descompresión central, osteotomía y reemplazo articular.[51] También se han usado células madre mesenquimales autógenas
percutáneas para el tratamiento de la osteonecrosis de tobillo en niños y adolescentes sobrevivientes de leucemia.[53]
Los factores que aumentan el riesgo de osteonecrosis son los siguientes:
Exposición a corticoesteroides y, posiblemente, administración simultánea de metotrexato y asparaginasa
El factor de tratamiento más importante que confiere riesgo de osteonecrosis es la exposición prolongada a los corticoesteroides, que es característica en los regímenes usados para la LLA, el linfoma de Hodgkin, el linfoma no Hodgkin y el TCMH.[47-49,54-56]
Es posible que el riesgo de osteonecrosis se relacione con el tipo de corticoesteroide; en algunos estudios de pacientes de LLA se indica un mayor riesgo con el uso de dexametasona que con prednisona.[57,58]
El cronograma de dosificación de los corticoesteroides también afecta el riesgo de presentar osteonecrosis. En el ensayo del Children’s Oncology Group (COG)-1961 para la LLA de riesgo alto recién diagnosticada, los pacientes se asignaron al azar para recibir dexametasona continua (diaria) o un cronograma de semanas alternadas de dexametasona durante la fase de intensificación diferida; el cronograma de semanas alternadas se relacionó con una incidencia más baja de osteonecrosis.[42]
Además de la exposición a corticoesteroides, la exposición al metotrexato y la asparaginasa simultánea contribuyen a la presentación de osteonecrosis.
Presentación de tromboembolismo durante el tratamiento antileucémico
En una revisión retrospectiva de 208 niños tratados por LLA, los investigadores de la McMaster University notificaron un aumento de 5,21 veces (IC 95 %, 1,82–14,91) de las probabilidades de osteonecrosis entre los niños que experimentaron tromboembolismo durante el tratamiento antileucémico, en comparación con los que no presentaban un tromboembolismo, incluso después de tener en cuenta la edad y la exposición a la asparaginasa.[59]
Acondicionamiento para un trasplante de células madre hematopoyéticas y evolución
En un estudio grande de casos y controles en el que se evaluaron los factores de riesgo para la osteonecrosis mediante el uso de datos del Center for International Blood and Marrow Transplant Research, se observaron riesgos más bajos de osteonecrosis en pacientes con enfermedades benignas y en quienes recibieron regímenes de acondicionamiento de intensidad reducida para enfermedades malignas, en comparación con los observados en los pacientes que recibieron regímenes mielosupresores para enfermedades malignas.[60]
En varios estudios se notificó un aumento del riesgo de osteonecrosis relacionado con la enfermedad de injerto contra huésped (EICH) crónica.[45,54,60]
Edad en el momento del diagnóstico o el trasplante
En varios estudios se demostró que la edad en el momento del diagnóstico (o en el momento del trasplante) es un factor de predicción independiente significativo de osteonecrosis.[42,47,57]
La osteonecrosis es significativamente más común en los niños mayores y los adolescentes que en los niños más pequeños. En el ensayo COG-1961 para la LLA de riesgo alto, la incidencia acumulada a 5 años de osteonecrosis sintomática fue del 1,0 % para los pacientes de 1 a 9 años, del 9,9 % para los pacientes de 10 a 15 años y del 20 % para los pacientes de 16 a 21 años (P < 0,0001).[42]
Raza
La osteonecrosis es más frecuente en pacientes blancos que en pacientes negros.[55,61]
Factores genéticos
Los factores genéticos que influyen en el metabolismo de los folatos, el metabolismo de los glucocorticoides y la adipogénesis se relacionaron con un exceso de riesgo de osteonecrosis en los sobrevivientes.[55,62,63]
En 2 estudios de genes propuestos se indican que los niños homocigóticos para una repetición de 28 pares de bases dentro de la región 5' no traducida del gen TYMS , tienen un mayor riesgo de osteonecrosis.[55,63] Este gen se vincula con la producción y el reemplazo de folato, y se inhibe con metotrexato.
Los investigadores del St. Jude Children's Research Hospital observaron un riesgo casi 6 veces más alto (OR, 5,6; IC 95 %, 2,7–11,3) de osteonecrosis en los sobrevivientes con polimorfismos del gen ACP1, que regula las concentraciones de lípidos y la diferenciación de los osteoblastos.[46]
En los estudios de asociación del genoma completo, se identificaron posibles variantes de riesgo del gen BMP7, la fusión PROX1::AS1, y los genes GRID2 (en niños menores de 10 años) y GRIN3A, todos los cuales se vinculan con actividad del receptor de glucocorticoides.[62,64]
Sexo
Los estudios que evaluaron la influencia del sexo en el riesgo de osteonecrosis produjeron resultados contradictorios, aunque algunos indican una incidencia más alta en las mujeres [51,61,65] que no fue confirmada por otros estudios.[66]
Adolescentes y adultos jóvenes
Se evaluaron las comorbilidades crónicas en una cohorte de 6778 sobrevivientes de 2 años de cáncer en adolescentes y adultos jóvenes (AAJ) (definidos como personas diagnosticadas de cáncer entre los 15 y los 39 años de edad), con una mediana de seguimiento de 5,1 años tras el diagnóstico de cáncer. El riesgo de presentar comorbilidades crónicas se comparó con el de personas emparejadas sin antecedentes de cáncer. La tasa de incidencia (TIR) de todas las comorbilidades fue más elevada en el caso de la osteonecrosis (TIR, 8,3), seguida de la osteoporosis (TIR, 5,75) y la artroplastia (TIR, 3,89). El uso de metotrexato (TIR, 21,6 para cualquier dosis) y corticosteroides (TIR, 5,4 para cualquier dosis) se asoció de forma significativa con la osteonecrosis.[67]
Osteocondroma
Los osteocondromas son protuberancias óseas benignas que se presentan de forma espontánea o en relación con la radioterapia. Por lo general, se presentan como una lesión única; sin embargo, a veces hay lesiones múltiples en el contexto de una osteocondromatosis hereditaria múltiple.[68]
La terapia con hormona del crecimiento tal vez influya en el momento de la presentación y en el ritmo de crecimiento de los osteocondromas.[14,69]
Dado que la degeneración maligna de estas lesiones es muy infrecuente, es más apropiado el seguimiento clínico que el radiológico.[70]
La resección quirúrgica solo es necesaria cuando la lesión interfiere con el alineamiento articular y el movimiento.[71]
Evidencia (riesgo de osteocondroma):
Cerca del 5 % de los niños sometidos a TCMH con mielosupresión presentarán un osteocondroma, por lo común, en las regiones metafisarias de los huesos largos.[68,72]
En un estudio italiano grande, se notificó un riesgo acumulado del 6,1 % de aparición de osteocondroma 15 años después del trasplante, con un aumento del riesgo relacionado con una edad más temprana en el momento del trasplante (≤3 años) y el uso de ICT.[73]
Se han notificado osteocondromas en pacientes con neuroblastoma que recibieron radioterapia local, terapia con anticuerpos monoclonales anti-GD2 e isotretinoína.[74]
Los osteocondromas se presentaron en el momento de una mediana de 8,2 años a partir del diagnóstico y la tasa de incidencia acumulada fue del 4,9 % a los 10 años del diagnóstico en 362 pacientes menores de 10 años.
En esta serie, la mayoría de los osteocondromas no se relacionaron con radiación y presentaron características de osteocondroma benigno del desarrollo.
Todavía son objeto de especulación la función patógena de la quimioterapia, la terapia con anticuerpos monoclonales anti-GD2 y la isotretinoína en la formación de un osteocondroma.
Amputación y cirugía con conservación del miembro
La amputación y la cirugía con conservación de miembro previenen la recidiva local de tumores de hueso al extirpar toda la enfermedad macroscópica y microscópica. Si se realizan de modo óptimo, ambos procedimientos logran la extirpación en bloque del tumor con un margen de tejido normal no afectado.
El tipo de procedimiento quirúrgico, el sitio del tumor primario y la edad del paciente afectan al riesgo de complicaciones posquirúrgicas.[75]
Las complicaciones en los sobrevivientes tratados con amputación incluyen problemas de ajuste de la prótesis, dolor crónico en el otro miembro, dolor en el miembro fantasma e hipertrofia ósea.[75,76]
Si bien las cirugías con conservación del miembro ofrecen un desenlace estéticamente más agradable, se notificaron complicaciones más frecuentes en sobrevivientes sometidos a estos procedimientos que en aquellos tratados con amputación.[75,77]
Las complicaciones posteriores a una cirugía con conservación del miembro incluyen falta de unión, fracturas patológicas, aflojamiento aséptico, discrepancias en la longitud de las extremidades, fracturas de las endoprótesis y amplitud limitada del movimiento articular.[75,77]
En ocasiones, se presentan complicaciones resistentes al tratamiento después de una cirugía con conservación del miembro que exigen una amputación.[78,79]
En varios estudios se compararon los desenlaces funcionales después de la amputación y la cirugía con conservación del miembro, pero los resultados fueron limitados debido al uso de métodos heterogéneos de evaluación funcional y de cohortes de tamaño pequeño.
En términos generales, los datos indican que la cirugía con conservación del miembro produce un funcionamiento mejor que la amputación, pero las diferencias son relativamente modestas.[75,79,80]
De manera similar, no hay una diferencia considerable entre los desenlaces en términos de calidad de vida a largo plazo en los sobrevivientes sometidos a amputación y los procedimientos quirúrgicos con conservación del miembro.[78]
En un análisis longitudinal del estado de salud en sobrevivientes de sarcoma de las extremidades en el CCSS, se indica una relación entre la amputación de extremidades inferiores y el aumento de las limitaciones en la actividad con la edad, así como una relación entre la amputación de la extremidad superior y menores logros académicos.[81]
Los investigadores del CCSS evaluaron los factores de riesgo y los desenlaces de la amputación tardía en los sobrevivientes que recibieron tratamiento para sarcomas de extremidades inferiores.[82]
La incidencia acumulada de amputación a los 25 años del diagnóstico de cáncer y la cirugía primaria con conservación del miembro fue del 18 % y no difirió significativamente entre los sobrevivientes diagnosticados en los años setenta (27 %) versus los años ochenta (19 %) y los años noventa (15 %).
Los factores relacionados con un aumento del riesgo de amputación tardía fueron el sexo masculino (cociente de riesgos instantáneos [CRI], 2,2; IC 95 %, 1,07–3,82) y los antecedentes de reconstrucción protésica de la articulación (CRI, 2,58; IC 95 %, 1,37–4,84).
Los sobrevivientes tratados con amputación primaria y aquellos que experimentaron amputación tardía después de una cirugía con conservación del miembro tuvieron más probabilidades de informar de un funcionamiento físico deficiente (RR, 1,31; IC 95 %, 1,04–1,72 y RR, 2,46; IC 95 %, 1,66–3,63, respectivamente) y puntajes de dolor corporal más precarios (RR, 1,50; IC 95 %, 1,10–2,06 y RR, 2,33; IC 95 %, 1,27–4,26, respectivamente), en comparación con aquellos sin amputación después de una cirugía con conservación del miembro.
Se evaluó la incidencia de intervenciones quirúrgicas importantes tardías entre los sobrevivientes de cáncer infantil a través de los datos del CCSS. En el informe, los sobrevivientes de sarcoma de Ewing y osteosarcoma presentaron la mayor carga acumulada de intervenciones quirúrgicas importantes tardías entre todos los sobrevivientes de tumores sólidos (la media acumulada de intervenciones quirúrgicas tardías fue de 322,9 por 100 sobrevivientes de sarcoma de Ewing y de 269,6 por 100 sobrevivientes de osteosarcoma). Un gran componente de esta carga está relacionado con el aumento de la tasa de cirugías musculoesqueléticas tardías adicionales, como artroplastia, amputación, revisión protésica causada por una infección, fallo del dispositivo o fracturas relacionadas. La cirugía locorregional o los tratamientos oncológicos con radioterapia se asociaron con la realización de intervenciones quirúrgicas tardías en la misma región corporal o sistema orgánico.[83]
Contracturas articulares
El TCMH con cualquier antecedente de EICH crónica se relaciona con contracturas articulares.[84,85]
En el Cuadro 15 se resumen los efectos tardíos óseos y articulares, y los exámenes médicos de detección correspondientes.
Cuadro 15. Efectos tardíos óseos y articularesa
Tratamiento predisponente
Efectos osteomusculares
Exámenes médicos de detección
TC = tomografía computarizada; DXA = absorciometría dual de rayos X; EICH = enfermedad de injerto contra huésped; TCMH = trasplante de células madre hematopoyéticas.
Radioterapia con exposición del sistema osteomuscular
Hipoplasia, fibrosis, crecimiento reducido o irregular (escoliosis, cifosis) y discrepancia en la longitud de las extremidades
Hallazgos del examen: huesos y tejidos blandos de los campos de radiación
Radioterapia con exposición de la cabeza y el cuello
Anomalías craneofaciales
Antecedentes: evaluación psicosocial con atención especial al progreso educativo o vocacional, depresión, ansiedad, estrés postraumático y aislamiento social
Examen de la cabeza y el cuello
Radioterapia con exposición del sistema osteomuscular
Fracturas inducidas por la radiación
Examen del hueso afectado
Metotrexato, corticoesteroides (dexametasona y prednisona), radioterapia con exposición del sistema osteomuscular y TCMH
Reducción de la densidad mineral ósea
Prueba de densidad mineral ósea (DXA o TC cuantitativa)
Corticoesteroides (dexametasona, prednisona)
Osteonecrosis
Antecedentes: dolor articular, hinchazón, inmovilidad y amplitud limitada de movimientos
Examen osteomuscular
Radiación con exposición de la cavidad oral
Osteorradionecrosis
Antecedentes y examen oral: alteración o retraso de la cicatrización después de un tratamiento dental, dolor en la mandíbula o hinchazón persistentes, y trismo
Amputación
Complicaciones relacionadas con la amputación (alteraciones cosméticas, limitaciones funcionales o de la actividad, integridad del muñón, dolor crónico y aumento de gasto energético)
Antecedentes: dolor, limitaciones funcionales o de la actividad
Hallazgos del examen: integridad del muñón
Evaluación de la prótesis
Cirugía con conservación del miembro
Complicaciones de la cirugía con conservación del miembro (limitaciones funcionales o de la actividad, fibrosis, contracturas, infecciones crónicas, discrepancia en la longitud de las extremidades, aumento del gasto energético y disfunción de la prótesis [aflojamiento, falta de unión y fracturas])
Antecedentes: dolor, limitaciones funcionales o de la actividad
Estilo CL, Huryn JM, Kraus DH, et al.: Effects of therapy on dentofacial development in long-term survivors of head and neck rhabdomyosarcoma: the memorial sloan-kettering cancer center experience. J Pediatr Hematol Oncol 25 (3): 215-22, 2003. [PUBMED Abstract]
Gevorgyan A, La Scala GC, Neligan PC, et al.: Radiation-induced craniofacial bone growth disturbances. J Craniofac Surg 18 (5): 1001-7, 2007. [PUBMED Abstract]
Karsila-Tenovuo S, Jahnukainen K, Peltomäki T, et al.: Disturbances in craniofacial morphology in children treated for solid tumors. Oral Oncol 37 (7): 586-92, 2001. [PUBMED Abstract]
Choi SY, Kim MS, Yoo S, et al.: Long term follow-up results of external beam radiotherapy as primary treatment for retinoblastoma. J Korean Med Sci 25 (4): 546-51, 2010. [PUBMED Abstract]
Shildkrot Y, Kirzhner M, Haik BG, et al.: The effect of cancer therapies on pediatric anophthalmic sockets. Ophthalmology 118 (12): 2480-6, 2011. [PUBMED Abstract]
Brownstein CM, Mertens AC, Mitby PA, et al.: Factors that affect final height and change in height standard deviation scores in survivors of childhood cancer treated with growth hormone: a report from the childhood cancer survivor study. J Clin Endocrinol Metab 89 (9): 4422-7, 2004. [PUBMED Abstract]
Nandagopal R, Laverdière C, Mulrooney D, et al.: Endocrine late effects of childhood cancer therapy: a report from the Children's Oncology Group. Horm Res 69 (2): 65-74, 2008. [PUBMED Abstract]
Sklar CA, Constine LS: Chronic neuroendocrinological sequelae of radiation therapy. Int J Radiat Oncol Biol Phys 31 (5): 1113-21, 1995. [PUBMED Abstract]
Packer RJ, Boyett JM, Janss AJ, et al.: Growth hormone replacement therapy in children with medulloblastoma: use and effect on tumor control. J Clin Oncol 19 (2): 480-7, 2001. [PUBMED Abstract]
Chow EJ, Friedman DL, Yasui Y, et al.: Decreased adult height in survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. J Pediatr 150 (4): 370-5, 375.e1, 2007. [PUBMED Abstract]
Bongers ME, Francken AB, Rouwé C, et al.: Reduction of adult height in childhood acute lymphoblastic leukemia survivors after prophylactic cranial irradiation. Pediatr Blood Cancer 45 (2): 139-43, 2005. [PUBMED Abstract]
Leung W, Ahn H, Rose SR, et al.: A prospective cohort study of late sequelae of pediatric allogeneic hematopoietic stem cell transplantation. Medicine (Baltimore) 86 (4): 215-24, 2007. [PUBMED Abstract]
Sanders JE: Growth and development after hematopoietic cell transplant in children. Bone Marrow Transplant 41 (2): 223-7, 2008. [PUBMED Abstract]
Sanders JE, Guthrie KA, Hoffmeister PA, et al.: Final adult height of patients who received hematopoietic cell transplantation in childhood. Blood 105 (3): 1348-54, 2005. [PUBMED Abstract]
Shalitin S, Laur E, Lebenthal Y, et al.: Endocrine complications and components of the metabolic syndrome in survivors of childhood malignant non-brain solid tumors. Horm Res Paediatr 81 (1): 32-42, 2014. [PUBMED Abstract]
Silber JH, Littman PS, Meadows AT: Stature loss following skeletal irradiation for childhood cancer. J Clin Oncol 8 (2): 304-12, 1990. [PUBMED Abstract]
Hartley KA, Li C, Laningham FH, et al.: Vertebral body growth after craniospinal irradiation. Int J Radiat Oncol Biol Phys 70 (5): 1343-9, 2008. [PUBMED Abstract]
Paulino AC, Nguyen TX, Mai WY: An analysis of primary site control and late effects according to local control modality in non-metastatic Ewing sarcoma. Pediatr Blood Cancer 48 (4): 423-9, 2007. [PUBMED Abstract]
Oshiro Y, Mizumoto M, Pan H, et al.: Spinal changes after craniospinal irradiation in pediatric patients. Pediatr Blood Cancer 67 (12): e28728, 2020. [PUBMED Abstract]
Hogeboom CJ, Grosser SC, Guthrie KA, et al.: Stature loss following treatment for Wilms tumor. Med Pediatr Oncol 36 (2): 295-304, 2001. [PUBMED Abstract]
Paulino AC, Wen BC, Brown CK, et al.: Late effects in children treated with radiation therapy for Wilms' tumor. Int J Radiat Oncol Biol Phys 46 (5): 1239-46, 2000. [PUBMED Abstract]
Fiscaletti M, Samoilenko M, Dubois J, et al.: Predictors of Vertebral Deformity in Long-Term Survivors of Childhood Acute Lymphoblastic Leukemia: The PETALE Study. J Clin Endocrinol Metab 106 (2): 512-525, 2021. [PUBMED Abstract]
Wilson CL, Dilley K, Ness KK, et al.: Fractures among long-term survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. Cancer 118 (23): 5920-8, 2012. [PUBMED Abstract]
Wilson CL, Chemaitilly W, Jones KE, et al.: Modifiable Factors Associated With Aging Phenotypes Among Adult Survivors of Childhood Acute Lymphoblastic Leukemia. J Clin Oncol 34 (21): 2509-15, 2016. [PUBMED Abstract]
van Atteveld JE, de Winter DTC, Pluimakers VG, et al.: Risk and determinants of low and very low bone mineral density and fractures in a national cohort of Dutch adult childhood cancer survivors (DCCSS-LATER): a cross-sectional study. Lancet Diabetes Endocrinol 11 (1): 21-32, 2023. [PUBMED Abstract]
van der Sluis IM, van den Heuvel-Eibrink MM, Hählen K, et al.: Altered bone mineral density and body composition, and increased fracture risk in childhood acute lymphoblastic leukemia. J Pediatr 141 (2): 204-10, 2002. [PUBMED Abstract]
Arikoski P, Komulainen J, Riikonen P, et al.: Reduced bone density at completion of chemotherapy for a malignancy. Arch Dis Child 80 (2): 143-8, 1999. [PUBMED Abstract]
Brennan BM, Mughal Z, Roberts SA, et al.: Bone mineral density in childhood survivors of acute lymphoblastic leukemia treated without cranial irradiation. J Clin Endocrinol Metab 90 (2): 689-94, 2005. [PUBMED Abstract]
Warner JT, Evans WD, Webb DK, et al.: Relative osteopenia after treatment for acute lymphoblastic leukemia. Pediatr Res 45 (4 Pt 1): 544-51, 1999. [PUBMED Abstract]
Gurney JG, Kaste SC, Liu W, et al.: Bone mineral density among long-term survivors of childhood acute lymphoblastic leukemia: results from the St. Jude Lifetime Cohort Study. Pediatr Blood Cancer 61 (7): 1270-6, 2014. [PUBMED Abstract]
den Hoed MA, Klap BC, te Winkel ML, et al.: Bone mineral density after childhood cancer in 346 long-term adult survivors of childhood cancer. Osteoporos Int 26 (2): 521-9, 2015. [PUBMED Abstract]
den Hoed MA, Pluijm SM, te Winkel ML, et al.: Aggravated bone density decline following symptomatic osteonecrosis in children with acute lymphoblastic leukemia. Haematologica 100 (12): 1564-70, 2015. [PUBMED Abstract]
Goodenough CG, Baedke JL, Delaney AM, et al.: Attributable Risk and Consequences of Bone Mineral Density Deficits in Childhood Cancer Survivors. JAMA Netw Open 8 (1): e2454069, 2025. [PUBMED Abstract]
Kvammen JA, Stensvold E, Godang K, et al.: Bone mineral density and nutrition in long-term survivors of childhood brain tumors. Clin Nutr ESPEN 50: 162-169, 2022. [PUBMED Abstract]
Le Meignen M, Auquier P, Barlogis V, et al.: Bone mineral density in adult survivors of childhood acute leukemia: impact of hematopoietic stem cell transplantation and other treatment modalities. Blood 118 (6): 1481-9, 2011. [PUBMED Abstract]
Kodama M, Komura H, Shimizu S, et al.: Efficacy of hormone therapy for osteoporosis in adolescent girls after hematopoietic stem cell transplantation: a longitudinal study. Fertil Steril 95 (2): 731-5, 2011. [PUBMED Abstract]
Bhandari R, Teh JB, Herrera C, et al.: Prevalence and risk factors for vitamin D deficiency in long-term childhood cancer survivors. Pediatr Blood Cancer 68 (7): e29048, 2021. [PUBMED Abstract]
Paulino AC: Late effects of radiotherapy for pediatric extremity sarcomas. Int J Radiat Oncol Biol Phys 60 (1): 265-74, 2004. [PUBMED Abstract]
van Atteveld JE, Pluijm SMF, Ness KK, et al.: Prediction of Low and Very Low Bone Mineral Density Among Adult Survivors of Childhood Cancer. J Clin Oncol 37 (25): 2217-2225, 2019. [PUBMED Abstract]
Mattano LA, Devidas M, Nachman JB, et al.: Effect of alternate-week versus continuous dexamethasone scheduling on the risk of osteonecrosis in paediatric patients with acute lymphoblastic leukaemia: results from the CCG-1961 randomised cohort trial. Lancet Oncol 13 (9): 906-15, 2012. [PUBMED Abstract]
Bürger B, Beier R, Zimmermann M, et al.: Osteonecrosis: a treatment related toxicity in childhood acute lymphoblastic leukemia (ALL)--experiences from trial ALL-BFM 95. Pediatr Blood Cancer 44 (3): 220-5, 2005. [PUBMED Abstract]
Karimova EJ, Rai SN, Howard SC, et al.: Femoral head osteonecrosis in pediatric and young adult patients with leukemia or lymphoma. J Clin Oncol 25 (12): 1525-31, 2007. [PUBMED Abstract]
Campbell S, Sun CL, Kurian S, et al.: Predictors of avascular necrosis of bone in long-term survivors of hematopoietic cell transplantation. Cancer 115 (18): 4127-35, 2009. [PUBMED Abstract]
Kawedia JD, Kaste SC, Pei D, et al.: Pharmacokinetic, pharmacodynamic, and pharmacogenetic determinants of osteonecrosis in children with acute lymphoblastic leukemia. Blood 117 (8): 2340-7; quiz 2556, 2011. [PUBMED Abstract]
Girard P, Auquier P, Barlogis V, et al.: Symptomatic osteonecrosis in childhood leukemia survivors: prevalence, risk factors and impact on quality of life in adulthood. Haematologica 98 (7): 1089-97, 2013. [PUBMED Abstract]
Ali N, Gohar S, Zaky I, et al.: Osteonecrosis in children with acute lymphoblastic leukemia: A report from Children's Cancer Hospital Egypt (CCHE). Pediatr Blood Cancer 66 (1): e27440, 2019. [PUBMED Abstract]
Karimova EJ, Wozniak A, Wu J, et al.: How does osteonecrosis about the knee progress in young patients with leukemia?: a 2- to 7-year study. Clin Orthop Relat Res 468 (9): 2454-9, 2010. [PUBMED Abstract]
Padhye B, Dalla-Pozza L, Little D, et al.: Incidence and outcome of osteonecrosis in children and adolescents after intensive therapy for acute lymphoblastic leukemia (ALL). Cancer Med 5 (5): 960-7, 2016. [PUBMED Abstract]
te Winkel ML, Pieters R, Hop WC, et al.: Prospective study on incidence, risk factors, and long-term outcome of osteonecrosis in pediatric acute lymphoblastic leukemia. J Clin Oncol 29 (31): 4143-50, 2011. [PUBMED Abstract]
DeFeo BM, Kaste SC, Li Z, et al.: Long-Term Functional Outcomes Among Childhood Survivors of Cancer Who Have a History of Osteonecrosis. Phys Ther 100 (3): 509-522, 2020. [PUBMED Abstract]
Hernigou P, Auregan JC, Dubory A, et al.: Ankle osteonecrosis in fifty-one children and adolescent's leukemia survivors: a prospective randomized study on percutaneous mesenchymal stem cells treatment. Int Orthop 45 (9): 2383-2393, 2021. [PUBMED Abstract]
Faraci M, Calevo MG, Lanino E, et al.: Osteonecrosis after allogeneic stem cell transplantation in childhood. A case-control study in Italy. Haematologica 91 (8): 1096-9, 2006. [PUBMED Abstract]
Relling MV, Yang W, Das S, et al.: Pharmacogenetic risk factors for osteonecrosis of the hip among children with leukemia. J Clin Oncol 22 (19): 3930-6, 2004. [PUBMED Abstract]
Giertz M, Aarnivala H, Wilk Michelsen S, et al.: Symptomatic osteonecrosis in children treated for Hodgkin lymphoma: A population-based study in Sweden, Finland, and Denmark. Pediatr Blood Cancer 71 (11): e31250, 2024. [PUBMED Abstract]
Vrooman LM, Stevenson KE, Supko JG, et al.: Postinduction dexamethasone and individualized dosing of Escherichia Coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol 31 (9): 1202-10, 2013. [PUBMED Abstract]
Hyakuna N, Shimomura Y, Watanabe A, et al.: Assessment of corticosteroid-induced osteonecrosis in children undergoing chemotherapy for acute lymphoblastic leukemia: a report from the Japanese Childhood Cancer and Leukemia Study Group. J Pediatr Hematol Oncol 36 (1): 22-9, 2014. [PUBMED Abstract]
Badhiwala JH, Nayiager T, Athale UH: The development of thromboembolism may increase the risk of osteonecrosis in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 62 (10): 1851-4, 2015. [PUBMED Abstract]
Li X, Brazauskas R, Wang Z, et al.: Avascular necrosis of bone after allogeneic hematopoietic cell transplantation in children and adolescents. Biol Blood Marrow Transplant 20 (4): 587-92, 2014. [PUBMED Abstract]
Mattano LA, Sather HN, Trigg ME, et al.: Osteonecrosis as a complication of treating acute lymphoblastic leukemia in children: a report from the Children's Cancer Group. J Clin Oncol 18 (18): 3262-72, 2000. [PUBMED Abstract]
Karol SE, Mattano LA, Yang W, et al.: Genetic risk factors for the development of osteonecrosis in children under age 10 treated for acute lymphoblastic leukemia. Blood 127 (5): 558-64, 2016. [PUBMED Abstract]
Finkelstein Y, Blonquist TM, Vijayanathan V, et al.: A thymidylate synthase polymorphism is associated with increased risk for bone toxicity among children treated for acute lymphoblastic leukemia. Pediatr Blood Cancer 64 (7): , 2017. [PUBMED Abstract]
Karol SE, Yang W, Van Driest SL, et al.: Genetics of glucocorticoid-associated osteonecrosis in children with acute lymphoblastic leukemia. Blood 126 (15): 1770-6, 2015. [PUBMED Abstract]
Aricò M, Boccalatte MF, Silvestri D, et al.: Osteonecrosis: An emerging complication of intensive chemotherapy for childhood acute lymphoblastic leukemia. Haematologica 88 (7): 747-53, 2003. [PUBMED Abstract]
Elmantaser M, Stewart G, Young D, et al.: Skeletal morbidity in children receiving chemotherapy for acute lymphoblastic leukaemia. Arch Dis Child 95 (10): 805-9, 2010. [PUBMED Abstract]
Chao C, Bhatia S, Xu L, et al.: Chronic Comorbidities Among Survivors of Adolescent and Young Adult Cancer. J Clin Oncol 38 (27): 3161-3174, 2020. [PUBMED Abstract]
Bordigoni P, Turello R, Clement L, et al.: Osteochondroma after pediatric hematopoietic stem cell transplantation: report of eight cases. Bone Marrow Transplant 29 (7): 611-4, 2002. [PUBMED Abstract]
Taitz J, Cohn RJ, White L, et al.: Osteochondroma after total body irradiation: an age-related complication. Pediatr Blood Cancer 42 (3): 225-9, 2004. [PUBMED Abstract]
King EA, Hanauer DA, Choi SW, et al.: Osteochondromas after radiation for pediatric malignancies: a role for expanded counseling for skeletal side effects. J Pediatr Orthop 34 (3): 331-5, 2014 Apr-May. [PUBMED Abstract]
Danner-Koptik K, Kletzel M, Dilley KJ: Exostoses as a long-term sequela after pediatric hematopoietic progenitor cell transplantation: potential causes and increase risk of secondary malignancies from Ann & Robert H. Lurie Children's Hospital of Chicago. Biol Blood Marrow Transplant 19 (8): 1267-70, 2013. [PUBMED Abstract]
Faraci M, Bagnasco F, Corti P, et al.: Osteochondroma after hematopoietic stem cell transplantation in childhood. An Italian study on behalf of the AIEOP-HSCT group. Biol Blood Marrow Transplant 15 (10): 1271-6, 2009. [PUBMED Abstract]
Kushner BH, Roberts SS, Friedman DN, et al.: Osteochondroma in long-term survivors of high-risk neuroblastoma. Cancer 121 (12): 2090-6, 2015. [PUBMED Abstract]
Nagarajan R, Neglia JP, Clohisy DR, et al.: Limb salvage and amputation in survivors of pediatric lower-extremity bone tumors: what are the long-term implications? J Clin Oncol 20 (22): 4493-501, 2002. [PUBMED Abstract]
Aulivola B, Hile CN, Hamdan AD, et al.: Major lower extremity amputation: outcome of a modern series. Arch Surg 139 (4): 395-9; discussion 399, 2004. [PUBMED Abstract]
Kaste SC, Neel MN, Rao BN, et al.: Complications of limb-sparing procedures using endoprosthetic replacements about the knee for pediatric skeletal sarcomas. Pediatr Radiol 31 (2): 62-71, 2001. [PUBMED Abstract]
Eiser C, Darlington AS, Stride CB, et al.: Quality of life implications as a consequence of surgery: limb salvage, primary and secondary amputation. Sarcoma 5 (4): 189-95, 2001. [PUBMED Abstract]
Renard AJ, Veth RP, Schreuder HW, et al.: Function and complications after ablative and limb-salvage therapy in lower extremity sarcoma of bone. J Surg Oncol 73 (4): 198-205, 2000. [PUBMED Abstract]
Fernandez-Pineda I, Hudson MM, Pappo AS, et al.: Long-term functional outcomes and quality of life in adult survivors of childhood extremity sarcomas: a report from the St. Jude Lifetime Cohort Study. J Cancer Surviv 11 (1): 1-12, 2017. [PUBMED Abstract]
Marina N, Hudson MM, Jones KE, et al.: Changes in health status among aging survivors of pediatric upper and lower extremity sarcoma: a report from the childhood cancer survivor study. Arch Phys Med Rehabil 94 (6): 1062-73, 2013. [PUBMED Abstract]
Geiger EJ, Liu W, Srivastava DK, et al.: What Are Risk Factors for and Outcomes of Late Amputation After Treatment for Lower Extremity Sarcoma: A Childhood Cancer Survivor Study Report. Clin Orthop Relat Res 481 (3): 526-538, 2023. [PUBMED Abstract]
Dieffenbach BV, Murphy AJ, Liu Q, et al.: Cumulative burden of late, major surgical intervention in survivors of childhood cancer: a report from the Childhood Cancer Survivor Study (CCSS) cohort. Lancet Oncol 24 (6): 691-700, 2023. [PUBMED Abstract]
Inamoto Y, Storer BE, Petersdorf EW, et al.: Incidence, risk factors, and outcomes of sclerosis in patients with chronic graft-versus-host disease. Blood 121 (25): 5098-103, 2013. [PUBMED Abstract]
Efectos tardíos en el aparato reproductor
La cirugía, la radioterapia o la quimioterapia que afecten de manera negativa cualquiera de los componentes del sistema hipotalamohipofisario o las gónadas a veces comprometen los desenlaces reproductivos en los sobrevivientes de cáncer infantil. La evidencia científica para este desenlace en los sobrevivientes de cáncer infantil es limitada debido a que los estudios se caracterizan por el tamaño pequeño de la muestra, el sesgo de selección y participación de la cohorte, la evaluación transversal, la heterogeneidad en el abordaje de tratamiento, el tiempo transcurrido desde el tratamiento y el método de confirmación. En particular, la bibliografía no ofrece resultados objetivos sobre la capacidad reproductiva (por ejemplo, análisis de semen en los hombres, recuento de folículos primordiales en las mujeres) y los desenlaces después de los abordajes contemporáneos de tratamiento adaptados al riesgo.[1,2]
El riesgo de esterilidad por lo general se relaciona con los tejidos u órganos comprometidos por el cáncer y el tipo específico de tratamiento citotóxico, la dosis y la combinación terapéutica.
La orquiectomía o la ooforectomía realizadas para el tratamiento de tumores de células germinativas en la niñez reducen la cantidad de células germinativas.
Los alquilantes y los fármacos similares que producen entrecruzamiento en las hebras del DNA que se usan en el tratamiento de los cánceres infantiles; son los fármacos quimioterapéuticos principales relacionados con un riesgo alto de esterilidad. Los factores que influyen en el riesgo de lesiones gonadales en niños tratados con quimioterapia de alquilantes son los siguientes:
Dosis acumulada.
En los estudios iniciales se usaron las dosis de alquilantes para definir las cantidades relacionada con riesgo de toxicidad gonadal en una cohorte específica. Los investigadores del Childhood Cancer Survivor Study (CCSS) crearon la dosis equivalente de ciclofosfamida como parámetro para normalizar las dosis acumuladas de varios alquilantes que es independiente de la población en estudio. La dosis de alquilante y la dosis equivalente de ciclofosfamida tienen un desempeño similar cuando se usan en varios modelos para diferentes desenlaces en los sobrevivientes que comprenden exposiciones de tratamiento, pero solo la dosis equivalente de ciclofosfamida permite una comparación entre cohortes sometidas a diferentes tratamientos. Las investigaciones en las que se evalúan los factores de riesgo de toxicidad gonadal varían en cuanto al uso de dosis acumuladas según cada alquilante, la dosis del alquilante y la dosis equivalente de ciclofosfamida.[3]
Alquilante específico.
Duración del tratamiento.
Edad en el momento del tratamiento.
Sexo.
El riesgo de lesión por radiación en el sistema hipotalamohipofisario o las gónadas se relaciona con el volumen tratado, la dosis total, el esquema de fraccionamiento y la edad en el momento del tratamiento.
El St. Jude Children's Research Hospital informó del riesgo acumulado de hipogonadismo e infertilidad en una cohorte de 156 pacientes pediátricos con meduloblastomas que recibieron tratamiento con cirugía, irradiación craneoespinal adaptada al riesgo y quimioterapia de dosis intensiva en el estudio SJMB03 (2003–2013).[4]
En toda la cohorte, la incidencia acumulada a 5 años de hipogonadismo fue del 33,9 %, y la incidencia acumulada a 10 años fue del 45,1 %.
La incidencia acumulada de hipogonadismo a 10 años fue del 82,4 % en las mujeres y del 18 % en los hombres (P < 0,0001).
La incidencia fue del 62,9 % para los pacientes que habían alcanzado la pubertad en el momento de la radioterapia y del 40,1 % para los que no la habían alcanzado (P = 0,0068).
La incidencia fue del 80,6 % para los que recibieron una dosis de radiación gonadal alta (>2 Gy), del 67,7 % para los que recibieron una dosis de radiación gonadal intermedia (1–2 Gy) y del 31,9 % para los que recibieron una dosis de radiación gonadal baja (<1 Gy) (P < 0,0001).
El hipogonadismo se relacionó con el sexo, el estado puberal, la exposición a la dosis de radiación gonadal, el intervalo desde el tratamiento hasta la pubertad y la dosis de vincristina.
La incidencia acumulada de infertilidad a 10 años fue del 55,4 % en las mujeres y del 23,5 % en los hombres (n = 33; P = 0,0389).
Además del tratamiento del cáncer, la edad en el momento del tratamiento y el sexo, es probable que los factores genéticos influyan en el riesgo de esterilidad permanente. Los protocolos pediátricos para el tratamiento del cáncer con frecuencia implican un tratamiento combinado; por lo tanto, quizá sea necesario considerar los efectos agregados de exposiciones gonadotóxicas en la evaluación de la capacidad reproductiva. Para calcular los riesgos de disfunción gonadal y esterilidad, se requiere de información detallada sobre las modalidades específicas de tratamiento del cáncer, incluso los procedimientos quirúrgicos específicos, el tipo y dosis acumuladas de fármacos quimioterapéuticos, y los volúmenes y las dosis de radioterapia.
El riesgo de esterilidad del tratamiento indicado no se traduce de manera automática en esterilidad durante la edad adulta. Esto es importante, en especial, para pacientes que no pudieron someterse a conservación de espermatozoides u ovocitos debido a una edad muy temprana en el momento del diagnóstico y tratamiento del cáncer.
En un estudio de sobrevivientes de la cohorte St. Jude Life (SJLIFE) (N = 1067), la mayoría de los adultos sobrevivientes de cáncer infantil (61,9 %) se consideraron vulnerables a la esterilidad, lo cual se asoció en forma marcada con factores sociodemográficos (edad más avanzada, etnia blanca, estar casado o tener pareja y estudios superiores), tratamiento gonadotóxico, inquietudes en torno a la capacidad reproductiva, intentos previos infructíferos para concebir y disfunción sexual (todas las P < 0,05).[5]
Las percepciones de riesgo a menudo fueron discordantes respecto del funcionamiento gonadal evaluado en el laboratorio. Se identificó deterioro de la función gonadal evaluada por laboratorio en 24 % de las sobrevivientes y 56% de los sobrevivientes varones, pero la concordancia con las percepciones de riesgo de los sobrevivientes fue baja. La mayoría de los sobrevivientes con percepciones discordantes sobrestimaron el riesgo (20 % de los sobrevivientes varones y 44 % de las sobrevivientes mujeres).
A los sobrevivientes en riesgo —como los que muestran signos iniciales de esterilidad y los que sufren incertidumbres, preocupación o malestar relacionados con la capacidad reproductiva— se les debe ofrecer pruebas de fertilidad.
Testículos
Los tratamientos del cáncer que tal vez perjudiquen el funcionamiento testicular y reproductivo son los siguientes:
Cirugía que afecta el funcionamiento de los testículos
Los pacientes sometidos a orquiectomía unilateral por torsión testicular a veces tienen un recuento subnormal de espermatozoides en el momento del seguimiento a largo plazo.[6,7]
La eyaculación retrógrada es una complicación frecuente de la disección ganglionar bilateral retroperitoneal en hombres con neoplasias testiculares.[8,9] No se dispone de desenlaces a largo plazo después de la disección ganglionar retroperitoneal mínimamente invasiva.[10]
La disfunción eréctil quizá se presente después de disecciones pélvicas extensas para extirpar un rabdomiosarcoma de próstata.[11,12]
Radiación que afecta el funcionamiento de los testículos
En los hombres tratados por cáncer infantil, existe la posibilidad de una lesión gonadal si los campos de tratamiento de la radiación incluyen la pelvis, las gónadas o todo el cuerpo.
El epitelio germinativo es más sensible a las lesiones por radiación que las células de Leydig que producen andrógenos.
A veces se observa una disminución en el recuento de espermatozoides entre 3 y 6 semanas después de la irradiación y, de acuerdo con la dosificación, la recuperación toma de 1 a 3 años.[13]
El epitelio germinativo se lesiona con dosificaciones mucho menores (<1 Gy) de radiación que las células de Leydig (20–30 Gy).[13]
Es posible que se produzca la insuficiencia irreversible de las células germinativas con dosis de más de 2 a 4 Gy de irradiación fraccionada.[13]
La administración de dosis más altas de radiación, como las de 24 Gy, que se usaron para tratar la recaída testicular de la leucemia linfoblástica aguda (LLA) conduce a la insuficiencia de las células germinativas y la disfunción de las células de Leydig.[14]
El daño que la radiación produce en las células de Leydig se relaciona con la dosis administrada y la edad en el momento del tratamiento.
La producción de testosterona tal vez sea normal en varones antes de la pubertad que reciben menos de 12 Gy de irradiación testicular fraccionada, pero las concentraciones plasmáticas elevadas de la hormona luteinizante observadas en este grupo indican una lesión subclínica.[15]
Por lo general, la insuficiencia gonadal se presenta cuando los varones prepúberes reciben más de 20 Gy de radiación dirigida a los testículos; se necesita la administración de terapia con andrógenos para lograr la masculinización.[15]
Se suele preservar la función de las células de Leydig en los varones sexualmente maduros si la dosis de radiación no excede los 30 Gy.[15]
Aunque los datos disponibles indican que las células de Leydig son más vulnerables cuando se exponen a la radiación antes de la pubertad, hay factores de confusión, como la edad en el momento de la prueba y los efectos de la orquiectomía y la quimioterapia, que limitan la confiabilidad de esta observación.[15]
Quimioterapia que afecta el funcionamiento de los testículos
La dosis acumulada del alquilante (por ejemplo, ciclofosfamida, clormetina o mecloretamina y dacarbazina) es un factor importante en el cálculo del riesgo de una lesión de células germinativas del testículo; sin embargo, se cuenta con una cantidad limitada de estudios que correlacionen los resultados del análisis del semen en cohortes bien caracterizadas clínicamente.[16]
En general, se conserva el funcionamiento de las células de Leydig, pero es común la insuficiencia de las células germinativas en hombres tratados con dosis acumuladas altas de ciclofosfamida (7500 mg/m2 o más) y más de 3 meses de terapia combinada con alquilantes.
En la mayoría de los estudios se indica que los varones prepúberes no experimentaron un riesgo más bajo de daño testicular inducido por la quimioterapia que los pacientes pospúberes.[17-20]
En los estudios de daño de las células germinativas testiculares, comprobado por oligospermia o azoospermia, después de la administración de alquilantes con radioterapia o sin esta, se notificó lo siguiente:
Ciclofosfamida:
Los varones sobrevivientes de linfoma no Hodgkin que recibieron una dosis acumulada de ciclofosfamida superior a 9,5 g/m2 y se sometieron a radioterapia pélvica tuvieron un aumento del riesgo de fracaso de recuperación de la espermatogénesis.[21]
En los sobrevivientes de sarcoma de Ewing y sarcoma de tejido blando, el tratamiento con una dosis acumulada de ciclofosfamida superior a 7,5 g/m2 se correlacionó con oligospermia o azoospermia persistentes.[22]
Las dosis de ciclofosfamida que excedieron los 7,5 g/m2 y las dosis de ifosfamida que excedieron los 60 g/m2 produjeron oligospermia o azoospermia en la mayoría de las personas expuestas.[23-25]
En un estudio de cohortes pequeño, se notificó una calidad normal del semen en adultos sobrevivientes a largo plazo de LLA infantil tratados con 0 a 10 g/m2 de ciclofosfamida e irradiación craneal, mientras que no se detectaron espermatozoides en muestras de semen de sobrevivientes tratados con más de 20 g/m2 de ciclofosfamida.[26]
El tratamiento con una dosis equivalente de ciclofosfamida inferior a 4 g/m2 produce, con poca frecuencia, azoospermia u oligospermia; aproximadamente un 89 % de los 31 hombres tratados tenían espermatozoides normales.[27]
De 15 hombres que recibieron 200 mg/kg de ciclofosfamida antes de someterse a un TCMH por anemia aplásica, el 67 % conservó la espermatogénesis.[28]
Dacarbazina:
La combinación de doxorrubicina, bleomicina, vinblastina y dacarbazina (ABVD) produjo a menudo oligospermia o azoospermia en adultos durante el curso de tratamiento. Sin embargo, recuperaron la espermatogénesis después de terminar el tratamiento, en contraste con la experiencia notificada después del tratamiento con clormetina o mecloretamina, vincristina, procarbazina y prednisona (MOPP).[29]
Alquilantes y procarbazina:
La mayoría de los estudios indican que la quimioterapia combinada con un alquilante y procarbazina produce un daño grave al epitelio germinativo del testículo que es irreversible cuando se administran dosis acumuladas altas.[17,30-33]
La azoospermia se presentó con menos frecuencia en adultos después del tratamiento con 2 ciclos de MOPP, en lugar de 6 ciclos de esta combinación.[34]
La elevación de la concentración inicial de la hormona foliculoestimulante (FSH), que refleja deterioro de la espermatogénesis, fue menos frecuente en los pacientes que recibieron 2 ciclos de vincristina, procarbazina, prednisona y doxorrubicina (OPPA) que en los que recibieron 2 ciclos de OPPA combinados con 2 o más ciclos de ciclofosfamida, vincristina, procarbazina y prednisona (COPP).[35]
Alquilantes y radiación craneal de dosis bajas:
En un estudio transversal, en el que se incluyeron adultos varones sobrevivientes de LLA infantil que recibieron quimioterapia de alquilantes con radiación craneal o sin esta, los investigadores del St. Jude Children's Research Hospital demostraron que la radiación craneal en dosis menores de 26 Gy no tiene ningún efecto comprobable independiente sobre la espermatogénesis.[36]
Funcionamiento testicular posterior a un trasplante de células madre hematopoyéticas
El riesgo de disfunción gonadal y esterilidad relacionada con el acondicionamiento con irradiación corporal total (ICT), dosis altas de quimioterapia con alquilantes, o ambos, es considerable.[37] Debido a que el trasplante a menudo se realiza para un cáncer recidivante o resistente al tratamiento, el tratamiento previo con quimioterapia con alquilantes o la radioterapia dirigida al sistema hipotalamohipofisario o a las gónadas quizás confiera riesgos adicionales.
En un estudio poblacional en el que se usó el National Quality Registry for Childhood Cancer de Suecia para evaluar la prevalencia y los factores de predicción de desenlaces endocrinos y reproductivos en jóvenes adultos sobrevivientes. En este estudio a escala nacional participaron 1212 hombres sobrevivientes de cáncer infantil con edades entre los 19 y los 40 años. De ellos, 99 se habían sometido a un TCMH con ICT o sin esta.[38]
Para los pacientes que se sometieron a TCMH (con ICT o sin esta), la prevalencia observada de pubertad inducida por hormonas fue del 11 %. La prevalencia de la terapia de reemplazo de testosterona (TRT) fue del 30,2 % y la prevalencia de tener hijos biológicos fue del 9,1 %.
Después del TCMH, las OR fueron de 4,5 para la pubertad inducida y de 13 para la TRT. La necesidad de recibir tratamiento para la pubertad inducida por hormonas y la TRT en curso se relacionaron de forma significativa con el TCMH (OR, 10,50, P < 0,001), en comparación con la ausencia del tratamiento para la pubertad inducida por hormonas o de la TRT en curso.
En un análisis longitudinal retrospectivo de los datos obtenidos durante un período de 30 años de una unidad de TCMH de atención terciaria, se incluyó a 130 hombres menores de 18 años en el momento en que se sometieron a un trasplante y que sobrevivieron 2 años o más después del TCMH. El 56 % de los pacientes eran prepúberes, mientras que el 44 % eran peripúberes
o pospúberes cuando se sometieron al TCMH.[39]
Los pacientes que eran prepúberes antes de someterse al TCMH tenían un menor riesgo de presentar hipogonadismo tras el TCMH (OR, 0,15), a pesar del menor volumen testicular general alcanzado en la edad adulta.
Los regímenes de quimioterapia sola se relacionaron con un volumen testicular estadísticamente mayor (P < 0,001), concentraciones más altas de testosterona (P = 0,008) y concentraciones más bajas de gonadotropina (P < 0,001) que los regímenes de ICT.
Se observó una disminución progresiva de la función de las células de Leydig a lo largo del tiempo debido a una tendencia a la baja de los concentraciones de testosterona después de alcanzar la pubertad plena, a pesar de un aumento compensatorio de las concentraciones de LH.
La exposición a ICT y busulfano se relacionó con la menor probabilidad de cursos sin episodios gonadales (P < 0,0001).
En un estudio francés de seguimiento a largo plazo de la leucemia en niños y adolescentes se evaluó la deficiencia de testosterona en varones sobrevivientes de leucemia infantil tras el tratamiento con TCMH o irradiación testicular.[37]
Entre los 178 pacientes tratados con ICT mieloablativa, el 55,6 % presentaba deficiencia total de testosterona, el 15,7 % tenía deficiencia parcial y el 28,7 % no tenía deficiencia. Un refuerzo testicular de 4 a 6 Gy de radioterapia y una edad más temprana en el momento del TCMH aumentaron de manera significativa el riesgo.
De los 53 pacientes que recibieron un régimen de acondicionamiento mieloablativo con busulfano, el 28,3 % presentaba deficiencia total, el 15,1 % tenía deficiencia parcial y el 56,6 % no tenía deficiencia. Entre los pacientes que recibieron este régimen y radiación testicular adicional, ninguno (0 %) fue normal, en comparación con un 62,5 % de los pacientes que no recibieron radiación testicular adicional.
El tratamiento con 24 Gy de radiación testicular sin TCMH indujo una deficiencia total o parcial en el 71,4 % y el 28,6 %, respectivamente (n = 21).
La edad en el momento del tratamiento también influye en el riesgo de lesiones gonadales. Los niños pequeños y los varones adolescentes tratados con dosis altas de ciclofosfamida (200 mg/kg), por lo general, conservarán el funcionamiento de las células de Leydig y la producción de testosterona, pero es común la insuficiencia de las células germinativas.[40]
Después del acondicionamiento con ICT, la mayoría de los varones conservan la capacidad de producir testosterona, pero tendrán insuficiencia de células germinativas.[40]
Hay datos limitados que permiten indicar que una proporción mayor de niños varones conservará la función germinativa o recuperará la espermatogénesis (de acuerdo con la evolución puberal y las concentraciones de gonadotropina) después del acondicionamiento de intensidad reducida con fludarabina y melfalán que aquellos tratados con acondicionamiento mielosupresor con busulfano y ciclofosfamida.[41]
Recuperación del funcionamiento de las células germinativas
Es posible recuperar el funcionamiento de las células germinativas después de la quimioterapia y la radioterapia citotóxicas; sin embargo, hay poca evidencia con desenlaces objetivos como el recuento espermático. En la mayoría de estudios se usan marcadores hormonales como las concentraciones de inhibina B y FSH para evaluar presencia de espermatogénesis; sin embargo, se notificaron limitaciones en la especificidad y el valor predictivo de un resultado positivo de estas pruebas.[42,43] Por lo tanto, se debe advertir a los varones sobrevivientes de que el análisis de semen es la evaluación más exacta para determinar la espermatogénesis adecuada.
Funcionamiento de las células de Leydig en sobrevivientes a largo plazo de cáncer infantil
El funcionamiento de las células de Leydig en los sobrevivientes de cáncer infantil no se ha estudiado de manera adecuada.
En un estudio poblacional, en el que se usó el National Quality Registry for Childhood Cancer de Suecia, 1212 hombres sobrevivientes de cáncer infantil con edades entre los 19 y los 40 años (mediana de edad en el momento del diagnóstico, 7 años; mediana de edad del estudio, 29 años; tratados entre 1981 y 2017) participaron en una encuesta autonotificada sobre tratamientos hormonales y la capacidad de engendrar un hijo biológico.[38]
En esta cohorte nacional, el 3,8 % de los sobrevivientes se había sometido a tratamiento hormonal para inducir la pubertad, el 6 % indicó que estaba recibiendo TRT y el 28,2 % notificó tener, al menos, un hijo biológico.
Los hombres que se habían sometido a radioterapia craneal o TCMH, así como los más jóvenes en el momento del diagnóstico, presentaron mayor probabilidad de someterse a pubertad inducida por hormonas y notificaron más TRT en curso. Los hombres que se habían sometido a un TCMH tenían 11 veces más probabilidades de seguir necesitando TRT después de la inducción.
Para la mayoría de los sobrevivientes que se sometieron a pubertad inducida por hormonas, la testosterona se combinó con la hormona del crecimiento. La mitad de los sobrevivientes que se sometieron a pubertad inducida por hormonas no estaban recibiendo TRT en la edad adulta joven y mostraron un pronóstico favorable para formar pareja y tener hijos biológicos.
Los hombres que estaban recibiendo TRT tenían una mayor prevalencia de otras deficiencias endocrinas (7–8 veces mayor) y a tomar medicamentos para el colesterol y el corazón, en comparación con los hombres que no recibían, TRT. Los hombres que necesitaban TRT tenían una menor probabilidad de vivir en pareja, ser padres o tener una ocupación actual. También notificaron una mayor fatiga y síntomas depresivos.
Los investigadores del estudio SJLIFE evaluaron la prevalencia y los factores de riesgo de la insuficiencia y la disfunción de las células de Leydig en 1516 hombres (mediana de edad, 30,8 años; mediana de tiempo desde el diagnóstico, 22 años).[44]
La prevalencia de la insuficiencia de las células de Leydig (definida como testosterona <250 ng/dl y hormona luteinizante >9,85 UI/l) fue del 6,9 %.
La prevalencia de la disfunción de las células de Leydig (definida como testosterona ≥250 ng/dl y hormona luteinizante >9,85 UI/L) fue de 14,7 %.
Los factores de riesgo independientes para la insuficiencia de las células de Leydig son una edad de 26 años o más en el momento de la evaluación, radioterapia testicular en cualquier dosis y alquilantes en dosis equivalentes a la ciclofosfamida de 4000 mg/m2 o más.
La orquiectomía unilateral y factores de riesgo similares para la insuficiencia de las células de Leydig se relacionaron con la disfunción de estas células.
El riesgo de insuficiencia y disfunción de las células de Leydig aumentó con la edad avanzada, las dosis de radioterapia más altas y el aumento de la exposición a alquilantes.
Los desenlaces adversos relacionados de manera significativa con la insuficiencia de las células de Leydig (pero no con su disfunción) son la obesidad abdominal, la diabetes mellitus, la disfunción eréctil, la debilidad muscular y la mortalidad por todas las causas.
Ovario
Los tratamientos del cáncer que perjudican el funcionamiento o la reserva ovárica son los siguientes:
El principal desafío para el cirujano pediátrico en el tratamiento de los tumores de ovario es encontrar el equilibrio adecuado entre la resección tumoral óptima y la preservación máxima de la fertilidad. La ooforectomía realizada para tratar los tumores de células germinativas reduce la reserva ovárica. En los tratamientos contemporáneos se utilizan procedimientos quirúrgicos para conservar la capacidad reproductiva combinados con quimioterapia sistémica con el fin de disminuir este riesgo.[45]
Radiación que afecta el funcionamiento ovárico
En las mujeres tratadas por cánceres infantiles, tal vez existe la posibilidad de lesión gonadal primaria si los campos de tratamiento incluyen la columna lumbosacra, el abdomen, la pelvis o todo el cuerpo. La frecuencia de insuficiencia ovárica después de la radioterapia abdominal se relaciona tanto con la edad de la paciente en el momento de la irradiación como con la dosis de radiación recibida por los ovarios. Los ovarios de las mujeres jóvenes son más resistentes al daño de la radiación que los de las mujeres de más edad por su mayor dotación de folículos primordiales.
La irradiación total del abdomen con dosis de 20 Gy o más se relaciona con el riesgo más alto de disfunción ovárica. De las pacientes en una serie, el 71 % no pudieron alcanzar la pubertad y el 26 % sufrió de menopausia prematura después de recibir dosis de irradiación total en el abdomen de 20 Gy a 30 Gy.[46] En otros estudios se notificaron resultados similares en mujeres tratadas con irradiación total del abdomen [47] o irradiación craneoespinal [48,49] durante la niñez.
Quimioterapia que afecta el funcionamiento ovárico
El funcionamiento ovárico a veces se altera después de un tratamiento con quimioterapia combinada que incluya un alquilante y procarbazina. En general, las niñas mantienen la funcionamiento gonadal al recibir dosis acumuladas de alquilantes más altas que los varones. La mayoría de las sobrevivientes de cáncer infantil tratadas con quimioterapia combinada adaptada al riesgo retienen o recuperan el funcionamiento ovárico. Sin embargo, el riesgo de insuficiencia ovárica aguda y menopausia prematura es considerable si el tratamiento incluye una modalidad combinada de quimioterapia con alquilantes y radioterapia abdominal o pélvica, o dosis intensivas de alquilantes para el acondicionamiento mielosupresor administrado antes de un TCMH.[50-55]
Insuficiencia ovárica prematura
La insuficiencia ovárica prematura está bien documentada en sobrevivientes de cáncer infantil; en particular, en mujeres tratadas con alquilantes y radioterapia abdominal.[50,54-57]
En los estudios, se relacionaron los siguientes factores con un aumento de la tasa de insuficiencia ovárica prematura (insuficiencia ovárica aguda y menopausia prematura):
Edad en el momento del tratamiento y edad alcanzada.
Aumento de las dosis de radioterapia abdominopélvica.
Exposición a alquilantes o procarbazina.
Ooforectomía.
El funcionamiento ovárico aparentemente normal al terminar la quimioterapia no se deberá interpretar como prueba de que no se produjo una lesión ovárica.
Evidencia (exceso de riesgo de insuficiencia ovárica prematura después de la administración de quimioterapia y radiación):
En un estudio del CCSS de 2008 sobrevivientes de tumor de Wilms no sindrómico unilateral, se encontró lo siguiente:[58]
La incidencia acumulada a 35 años de insuficiencia ovárica prematura fue del 7,3 % (intervalo de confianza [IC] 95 %, 5,0–9,7).
Los sobrevivientes del tumor de Wilms tratados con terapia más intensa (incluso radioterapia) presentaron tasas más altas de insuficiencia ovárica prematura, en comparación con los sobrevivientes tratados con vincristina y dactinomicina solos.
El estado menopáusico de 2930 sobrevivientes que participaron en un estudio del CCSS se comparó con el estado de 1399 hermanas. La menopausia prematura no quirúrgica se definió como el cese de la menstruación durante más de 6 meses que comienza 5 años después del diagnóstico del cáncer, pero antes de los 40 años, y que no tiene ninguna otra causa, como embarazo, cirugía o medicamentos.[54]
La prevalencia de menopausia prematura no quirúrgica fue del 9,1 % a los 40 años en estos sobrevivientes.
En los análisis multivariantes, los factores de riesgo independientes significativos de menopausia prematura no quirúrgica fueron los siguientes:
Exposición a dosis de procarbazina mayores a 4000 mg/m2 (oportunidad relativa [OR], 8,96; IC 95 %, 5,02–16,00; P < 0,0001).
Exposición a cualquier dosis de radioterapia dirigida a los ovarios (OR, 2,73 [IC 95 %, 1,33–5,61; P = 0,0062] para una dosis inferior a 5 Gy y OR, 8,02 [IC 95 %, 2,81–22,85; P < 0,0001] para una dosis superior a 5 Gy).
Ser receptor de TCMH (OR, 6,35; IC 95 %, 1,19–33,93; P = 0,0307).
Una dosis equivalente de ciclofosfamida de 6000 mg/m2 o más, que incluyó procarbazina fue significativa en el análisis univariante, pero no logró ser significativa en el análisis multivariante.
Para los sobrevivientes que recibieron dosis de procarbazina mayores de 4000 mg/m2 la prevalencia de menopausia prematura no quirúrgica a los 40 años fue del 39,7 % en comparación con el 4,2 % en las sobrevivientes que no recibieron procarbazina (P < 0,0001).
La exposición de los ovarios a una dosis de radiación mayor de 5 Gy produjo una prevalencia de menopausia prematura no quirúrgica a los 40 años del 24,1 %, en comparación con una prevalencia del 3,0 % para quienes no recibieron radiación (P < 0,0001).
La exposición a la ciclofosfamida (cualquier dosis), la ooforectomía unilateral, el hábito de fumar y el índice de masa corporal (IMC) no presentaron una relación significativa con el riesgo de menopausia prematura no quirúrgica.
En comparación con las sobrevivientes que no presentaron menopausia prematura no quirúrgica, aquellas que presentaron menopausia prematura no quirúrgica tuvieron una probabilidad más baja de embarazo o parto de un hijo vivo entre los 31 y los 40 años de edad. No se encontraron diferencias en las tasas de embarazos y recién nacidos vivos antes de los 30 años en sobrevivientes que finalmente presentaron menopausia prematura no quirúrgica y las que no presentaron este tipo de menopausia.
En un estudio francés de cohortes de 1109 mujeres sobrevivientes de cáncer sólido en la niñez, se identificaron los factores de riesgo de la menopausia no quirúrgica, que comprendieron el tratamiento con fármacos alquilantes, la radiación que expone los ovarios y la ooforectomía.[57]
Las mujeres tratadas con alquilantes después del inicio de la pubertad, ya fueran solos (riesgo relativo [RR], 9; IC 95 %, 2,7–28; P = 0,0003) o junto con dosis pequeñas de radiación dirigida a los ovarios (RR, 29, IC 95 %, 8–108; P < 0,0001), presentaron el razón de riesgos más alta de menopausia no quirúrgica.
La tasa general de menopausia no quirúrgica a los 40 años de edad fue de solo un 2,1 % y mucho más baja que la observada en los estudios de cohortes del CCSS y la European Organization for Research and Treatment of Cancer, que incluyeron a sobrevivientes de neoplasias malignas hematológicas.
La ooforectomía unilateral se relacionó con una menopausia 7 años más temprana.
Se encuestó a sobrevivientes europeas de linfoma de Hodgkin tratadas entre los 15 y 40 años, que no recibían anticonceptivos hormonales, sobre la presentación de insuficiencia ovárica prematura.[56]
En 460 mujeres, la insuficiencia ovárica prematura estuvo influida principalmente por el uso de quimioterapia con alquilantes con una relación lineal entre la dosis de quimioterapia con alquilantes y la aparición de insuficiencia ovárica prematura.
El riesgo de insuficiencia ovárica prematura aumentó un 23 % por año de edad en el momento del tratamiento. En las mujeres tratadas sin quimioterapia con alquilantes antes de los 32 años, el riesgo acumulado de insuficiencia ovárica prematura fue del 3 %, cuando el tratamiento se hizo a los 32 años o más, el riesgo fue del 9 %.
En los casos en que la menstruación regresó después del tratamiento, el riesgo acumulado de insuficiencia ovárica prematura fue independiente de la edad en el momento del tratamiento.
En las mujeres que en último término presentaron insuficiencia ovárica prematura, 22 % tuvo uno o más hijos después del tratamiento, comparadas con 41 % de las mujeres sin insuficiencia ovárica prematura que tuvieron uno o más hijos después del tratamiento.
Este informe indica que las mujeres fecundas después del tratamiento todavía pueden enfrentar problemas de esterilidad en una etapa posterior.
Los investigadores del estudio SJLIFE evaluaron la prevalencia de los factores de riesgo para la insuficiencia ovárica prematura en 921 sobrevivientes mujeres de cáncer infantil. La insuficiencia ovárica prematura se evaluó clínicamente y se definió por amenorrea persistente y una concentración de FSH de 30 UI/l o superior antes de los 40 años.[59]
La prevalencia de insuficiencia ovárica prematura fue del 10,9 % en las mujeres con una mediana de edad de 31,7 años en el momento de la evaluación del estudio y una mediana de 24 años transcurridos desde el diagnóstico de cáncer.
Los factores de riesgo independiente de insuficiencia ovárica prematura fueron la radioterapia ovárica de cualquier dosis y una dosis equivalente de ciclofosfamida de 8000 mg/m2 o más.
La obesidad (IMC de 30 kg/m2 o más) en el momento de la evaluación se relacionó con un riesgo más bajo de insuficiencia ovárica prematura (CRI, 0,36).
Las sobrevivientes con insuficiencia ovárica prematura tuvieron una probabilidad más alta de tener una densidad mineral ósea baja (OR, 5,07) y fragilidad (OR, 3,5) que las mujeres sin insuficiencia ovárica prematura.
Los datos sobre la función ovárica en sobrevivientes de neuroblastoma de riesgo alto son limitados. En una revisión retrospectiva se incluyó a 83 mujeres sobrevivientes de neuroblastoma de riesgo alto (mediana de edad, 19 años; mediana de seguimiento, 15 años) cuyo diagnóstico se había producido, al menos, 5 años atrás y que recibían seguimiento en un centro oncológico terciario. Se notificaron casos de disfunción ovárica, en particular tras un rescate autógeno de células madre.[60]
De las sobrevivientes, 49 (59 %) presentaron disfunción ovárica y 34 (41 %) presentaron insuficiencia ovárica prematura. La disfunción ovárica se definió como concentraciones de FSH de más de 15 mu/ml, y la insuficiencia ovárica prematura como disfunción ovárica persistente que requería terapia hormonal sustitutiva.
Las sobrevivientes tratadas con rescate de células madre autógenas (n = 51) tenían 3,2 veces más probabilidades de presentar disfunción ovárica (P < 0,001) y 4,5 veces más probabilidades de presentar insuficiencia ovárica prematura (P = 0,002), en comparación con las tratadas con quimioterapia convencional (n = 32), tras hacer el ajuste por edad alcanzada.
De las sobrevivientes, 2 del grupo de rescate de células madre no autógenas y 6 del grupo de rescate de células madre autógenas lograron, al menos, un embarazo espontáneo.
Funcionamiento ovárico posterior a un trasplante de células madre hematopoyéticas
La preservación del funcionamiento ovárico en las mujeres tratadas con un TCMH se relaciona con la edad en el momento del tratamiento, la administración de quimioterapia con alquilantes antes del trasplante y radioterapia abdominopélvica, así como el régimen de acondicionamiento para el trasplante.[52,61]
Evidencia (exceso de riesgo de insuficiencia ovárica prematura después de TCMH):
En un estudio observacional retrospectivo se incluyó a 178 mujeres sobrevivientes de leucemia infantil del programa francés de seguimiento a largo plazo. En el estudio se evaluaron mujeres que se habían sometido a TCMH prepuberal por leucemia aguda. Entre estas sobrevivientes, 116 habían recibido ICT y 62 habían recibido un protocolo de acondicionamiento a base de busulfano. La mediana de seguimiento desde el TCMH fue de 18 años.[62]
De 178 mujeres, 106 (60 %) requirieron inducción de la pubertad (con tratamiento hormonal sustitutivo), mientras que 72 (40 %) tuvieron menarquia espontánea.
Tras la menarquia espontánea, 33 pacientes (46 %) presentaron insuficiencia ovárica prematura, la mayoría en los 5 años siguientes al TCMH.
Los factores de riesgo de la insuficiencia ovárica prematura fueron la edad avanzada en el momento del TCMH y la criopreservación del tejido ovárico.
En las pacientes que se sometieron al TCMH antes de los 4,8 años de edad, más del 65 % tuvieron menarquia espontánea, y casi el 50 % no presentaron insuficiencia ovárica prematura.
En las pacientes que se sometieron a un TCMH después de los 10,9 años de edad, más del 85 % no tuvieron menarquia espontánea y necesitaron inducción de la pubertad (mediante terapia de reemplazo hormonal).
Las niñas y las mujeres jóvenes sometidas a acondicionamiento con ICT o regímenes con busulfano parecen tener el mismo riesgo alto de disminución del funcionamiento ovárico y menopausia prematura que las pacientes acondicionadas con ciclofosfamida sola.[52] Todas las mujeres que recibieron dosis altas (50 mg/kg/día x 4 días) de ciclofosfamida antes de un TCMH por anemia aplásica presentaron amenorrea después del trasplante.
En otra serie, 36 de 43 mujeres con anemia aplásica acondicionadas con ciclofosfamida (200 mg/kg) recuperaron el funcionamiento normal de los ovarios entre 3 y 42 meses después del trasplante, incluso las 27 pacientes que tenían entre 13 y 25 años en el momento del TCMH.[53]
La ICT es especialmente dañina cuando se administra en una sola fracción.[52] La mayoría de las mujeres pospúberes que reciben ICT antes de un TCMH presentan amenorrea.
En una serie, la recuperación del funcionamiento ovárico normal se presentó en solo 9 de 144 pacientes y se correlacionó de manera muy alta con la edad al momento de la radioterapia en el caso de pacientes menores de 25 años.[53]
Entre las mujeres con leucemia, la irradiación craneal antes del trasplante redujo aún más la posibilidad de conservar el funcionamiento ovárico.[52]
Es posible que el funcionamiento ovárico se conserve mejor (de acuerdo con el desarrollo puberal y las concentraciones de gonadotropina) en mujeres sometidas a TCMH con acondicionamiento de intensidad reducida con fludarabina y melfalán que en aquellas sometidas a acondicionamiento mielosupresor con busulfano y ciclofosfamida..[41]
Predicción de la insuficiencia ovárica prematura
Mediante el uso de datos del CCSS, los investigadores crearon modelos de predicción según el tratamiento para determinar los riesgos de las sobrevivientes de presentar insuficiencia ovárica primaria. Estos modelos proporcionan un riesgo específico para cada paciente en todo el espectro de edad reproductiva. Los modelos anteriores se utilizaron para determinar los riesgos de las sobrevivientes de presentar insuficiencia ovárica aguda en el transcurso de los 5 años siguientes al diagnóstico de cáncer.[63] La información obtenida a partir de estos modelos puede servir de base para los debates sobre la elección y el momento de los tratamientos para preservar la fertilidad. La cohorte SJLIFE se usó para validar los primeros modelos de predicción de riesgo específicos por edad para la insuficiencia ovárica primaria. Se incluyeron 7891 mujeres sobrevivientes del CCSS (922 con insuficiencia ovárica primaria) en la creación del modelo de predicción del riesgo de insuficiencia ovárica primaria y 1349 mujeres sobrevivientes del SJLIFE (101 con insuficiencia ovárica primaria) en el estudio de validación externa.[64]
La prevalencia estimada de insuficiencia ovárica primaria aumentó del 7,9 % al 18,6 % en el CCSS y del 7,3 % al 14,9 % en el SJLIFE a medida que se incrementaba la edad de las sobrevivientes de 21 a 40 años. Las pacientes que presentaron insuficiencia ovárica primaria, 531 de 922 (57,6 %) en el CCSS y 50 de 101 (49,5 %) en el SJLIFE, presentaron insuficiencia ovárica aguda.
El anterior modelo de predicción del riesgo (que se limitó al período de los 5 años posteriores al cáncer infantil) no fue útil para las sobrevivientes cuya menstruación se reanudó tras el tratamiento, pero que presentaron un mayor riesgo residual de menopausia prematura en el momento en que estaban preparadas para tener hijos. Este estudio abordó esta carencia.
Este modelo integral incorporó dosis acumuladas prescritas o administradas de radioterapia abdominal o pélvica, información de TCMH y una serie de fármacos de quimioterapia, en especial alquilantes individuales. El modelo funcionó bien en ambas cohortes de sobrevivientes.
En este estudio se evaluaron predictores genéticos, en concreto una puntuación de riesgo poligénica formada por variantes genéticas relacionadas con la insuficiencia ovárica primaria asociada con el tratamiento en sobrevivientes.
En un estudio multicéntrico neerlandés se evaluaron los factores del tratamiento que se asociaban con marcadores hormonales y ecográficos de reserva ovárica en sobrevivientes a 5 años de cánceres infantiles (diagnosticados entre 1963 y 2002) y en los controles. Entre 564 sobrevivientes y 390 controles sometidas a evaluación clínica de la reserva ovárica, incluso evaluación de la hormona antimuleriana (AMH), la FSH, la inhibina B y el recuento de folículos antrales (RFA).[65]
Entre los sobrevivientes de cáncer infantil, del 7,0 % al 17,7 % tenían marcadores de reserva ovárica normal en comparación con el 2,4 % al 13,6 % en los controles.
Se encontraron marcadores de reserva ovárica anormal en un número significativamente más alto de sobrevivientes de cáncer infantil que entre los controles (AMH: 26 vs. 4 %; RFA: 20 vs. 3 %; inhibina B: 42 vs. 16 %). Los valores de AMH y FSH también fueron significativamente diferentes en las sobrevivientes menores de 35 años.
Los tratamientos que exhibían una asociación independiente con por lo menos un marcador de reserva ovárica fueron la ciclofosfamida, la procarbazina, un grupo de otros alquilantes, la dactinomicina, la doxorrubicina, la mitoxantrona, la radioterapia vertebral, la radioterapia abdominopélvica y la ICT.
Solo se establecieron relaciones entre la dosis y el efecto para la procarbazina y la radioterapia abdominopélvica.
Fertilidad
La esterilidad sigue siendo uno de los efectos más comunes que alteran la vida de los sobrevivientes de cáncer infantil a largo plazo. Los estudios de cohortes de cáncer infantil demuestran el efecto del tratamiento citotóxico en los desenlaces reproductivos. Las investigaciones del CCSS dilucidaron los factores que contribuyen a la subfertilidad entre los sobrevivientes de cáncer infantil.[66,67]
No se identificaron anomalías en la fertilidad (características reproductivas y niveles de AMH, en comparación con los controles) en una serie de 56 mujeres sobrevivientes a largo plazo de cáncer de tiroides infantil diferenciado que recibieron yodo I 131 (131I) para el tratamiento. La mediana de seguimiento fue de 15,4 años (intervalo, 8,3–24,7 años), y la mediana de la dosis acumulada de 131I fue de 7,4 GBq/200,0 mCi. Ninguna de las sobrevivientes notificó menopausia prematura.[68]
Evidencia (exceso de riesgo de alteración de la fertilidad):
Se evaluó la fertilidad en 10 938 participantes del CCSS (5640 hombres, 5298 mujeres) y 3949 hermanos de ambos sexos.[66]
Al cabo de una mediana de seguimiento de 8 años desde el ingreso a la cohorte, el 38 % de los sobrevivientes informó haber estado en embarazo (mujeres) o haber engendrado un hijo (varones), lo que produjo al menos un recién nacido vivo en el 83 % de los sobrevivientes.
En los hermanos de ambos sexos sometidos a vigilancia durante una mediana de 10 años, el 62 % informó haber estado en embarazo (mujeres) o haber engendrado un hijo (varones), lo que produjo al menos un recién nacido vivo en el 90 % de los hermanos o hermanas.
En el análisis multivariante se confirmó que los sobrevivientes presentaron una reducción significativa en la probabilidad de engendrar un hijo o quedar embarazada (cociente de riesgos instantáneos [CRI], 0,63 en hombres y 0,87 en mujeres) o de tener un hijo vivo (CRI, 0,63 en hombres y 0,82 en mujeres) en comparación con los hermanos de ambos sexos.
Las dosis más altas de alquilantes (CRI, 0,82 por incrementos de 5000 mg/m2) y el cisplatino redujeron la probabilidad de engendrar en los hombres sobrevivientes, pero solo el busulfano y las dosis más altas (>411 mg/m2) de lomustina redujeron de manera significativa el embarazo en las mujeres.
El riesgo de tener una menor probabilidad de embarazo en las mujeres solo se observó con las dosis equivalentes más altas de ciclofosfamida (CRI, 0,85 para el cuartil más alto [≥11 295 mg/m2] vs. la ausencia de exposición).
Los CRI (IC 95 %) de la probabilidad de notificar el primer embarazo según la dosis equivalente de ciclofosfamida para hombres y mujeres sobrevivientes se resumen en el Cuadro 16:
Cuadro 16. Dosis equivalente de ciclofosfamida por tercil y sexo
Dosis equivalente de ciclofosfamida por tercil
Hombres
Mujeres
CRI (IC 95 %)
Valor de P
CRI (IC 95 %)
Valor de P
IC = intervalo de confianza; CRI = cociente de riesgos instantáneos.
Más baja (<4897 mg/m2)
1,14 (1,00–1,30)
0,045
0,97 (0,86–1,08)
0,55
Media (4897–9638 mg/m2)
0,79 (0,68–0,91)
0,0010
0,98 (0,87–1,11)
0,76
Alta (≥9639 mg/m2)
0,55 (0,47–0,64)
<0,0001
0,90 (0,79–1,01)
0,07
Se observaron relaciones semejantes en los desenlaces de recién nacidos vivos.
En ocasiones, la fertilidad se altera por factores distintos a la ausencia de espermatozoides u óvulos. La concepción exige el paso de espermatozoides por el cuello uterino, permeabilidad de las trompas de Falopio para que ocurra la fecundación, así como condiciones apropiadas para la implantación en el útero.
La eyaculación retrógrada sucede con una frecuencia significativa en hombres sometidos a una disección ganglionar retroperitoneal bilateral.[8,9]
La irradiación abdominal en ocasiones afecta la estructura del útero. En un estudio se demostró que la longitud del útero era significativamente más corta en 10 mujeres con insuficiencia ovárica tratadas con irradiación total del abdomen. El grosor endometrial no aumentó en respuesta a la terapia de reemplazo hormonal en 3 mujeres sometidas a un examen ecográfico semanal. En la ecografía Doppler, no se detectó flujo sanguíneo en ninguna de las arterias uterinas de 5 mujeres, ni en 1 de las arterias uterinas de 3 mujeres más.[69]
En un estudio sobre el estado menopáusico y los desenlaces reproductivos en 2930 sobrevivientes del CCSS, los investigadores encontraron que las mujeres que presentaron menopausia prematura no quirúrgica tuvieron tasas de embarazo y recién nacidos vivos mucho más bajas antes de la menopausia prematura no quirúrgica en el intervalo de edad de 31 a 40 años. Sin embargo, las tasas de embarazo y recién nacidos vivos no fueron diferentes durante el intervalo de edad de 21 a 30 años, de acuerdo con el estado menopáusico.[54]
En análisis multivariantes, las variables de tratamiento significativas para la menopausia prematura no quirúrgica fueron la exposición a la procarbazina en dosis mayores de 4000 mg2, cualquier dosis de irradiación ovárica y el TCMH.
Una dosis equivalente de ciclofosfamida de 6000 mg/m2 o más, que incluyó procarbazina fue significativa en el análisis univariante, pero no logró ser significativa en el análisis multivariante.
Funcionamiento sexual
No se ha estudiado a profundidad la salud psicosexual de los adultos tratados por cáncer durante la infancia, la adolescencia y la adultez temprana.
En un análisis del estudio SJLIFE se estimó la prevalencia y los factores de riesgo de disfunción sexual en 936 mujeres adultas sobrevivientes de cáncer infantil. En el estudio también se evaluaron las asociaciones entre la disfunción sexual y los síntomas psicológicos o la calidad de vida.[70]
La disfunción sexual fue prevalente en casi el 20 % de las sobrevivientes; las sobrevivientes de tumores de células germinativas (OR, 8,8), tumores renales (OR, 4,5) o leucemia (OR, 3,1) tenían un riesgo más alto que los controles.
Una mayor edad en el momento del seguimiento (45–54 vs. 18–24 años; OR, 5,7) y los antecedentes de cirugía pélvica (OR, 2,0), depresión (OR, 2,0), e hipogonadismo que recibían reemplazo hormonal (OR, 3,3) fueron factores vinculados con disfunción sexual.
Solo el 2,9 % de las sobrevivientes con disfunción sexual notificaron que recibían alguna intervención.
En un estudio prospectivo de cohortes (Project REACH), 249 sobrevivientes adultas de cáncer infantil (de 18 a 65 años) completaron mediciones de actividad física y salud mental, incluso disfunción sexual. El 32 % de las sobrevivientes experimentaron una disfunción sexual clínicamente significativa. Los problemas de salud física y mental, como la fatiga, la falta de sueño, el dolor, la salud física precaria, la salud mental precaria o la depresión, se relacionaron con la disfunción sexual y los antecedentes de un tumor del SNC.[71]
Reproducción
Conservación de la fertilidad
El progreso de la endocrinología reproductiva ha generado varias opciones para conservar la capacidad reproductiva o permitir la reproducción por otros métodos para las pacientes que están por recibir quimioterapia o radioterapia con potencial tóxico.[72-75]
Para los varones, la criopreservación de los espermatozoides antes del tratamiento es un método eficaz para eludir el efecto esterilizador del mismo para todos los varones peripúberes y pospúberes. Aunque se observó que la calidad del semen previa al tratamiento en los pacientes de cáncer es menor que la que se observa en donantes sanos, la disminución porcentual de la calidad del semen y el efecto dañino de la criopreservación en los espermatozoides de los pacientes de cáncer son similares en los donantes sanos.[76,77]
Para quienes no es posible almacenar su semen en bancos, las tecnologías más nuevas como la extracción de espermatozoides del testículo quizá sean una opción. Aún más, es probable que al usar los adelantos tecnológicos de micromanipulación, como la inyección intracitoplasmática de espermatozoides y técnicas similares, se logre una fecundación exitosa con espermatozoides extraídos quirúrgicamente o incluso con espermatozoides crioconservados de calidad precaria de los pacientes de cáncer.[78,79]
Según los registros nacionales de Noruega (Cancer Registry of Norway y Medical Birth Registry), se notificó que los sobrevivientes de cáncer varones jóvenes tenían un riesgo tres veces mayor de usar tecnología de reproducción asistida para la concepción de descendientes.[80]
En un estudio poblacional a nivel nacional, en el que se utilizó el Swedish Medical Birth Registry, los sobrevivientes de cáncer infantil y los sobrevivientes de cáncer adolescentes y adultos jóvenes tuvieron más probabilidad de lograr ser padres mediante inyección intracitoplasmática de espermatozoides que con la fertilización in vitro.[81]
Para las mujeres, las técnicas de reproducción asistida más exitosas dependen de la recolección y almacenamiento de los ovocitos durante el periodo pospuberal y la criopreservación de ovocitos no fecundados o embriones antes de la terapia gonadotóxica.[82]
Las opciones establecidas para las mujeres comprenden la transposición ovárica, la protección de la radiación y la crioconservación de ovocitos o embriones. En los Estados Unidos, la criopreservación de tejido ovárico se considera experimental en la actualidad, pero se realiza como un procedimiento de conservación de la fertilidad establecido en partes de Europa y en Israel.[83,84] Las opciones para las mujeres prepúberes se limitan a la crioconservación de tejido ovárico para un autotrasplante posterior, pero está en fase de investigación y se puede ofrecer a las niñas con cánceres que no son hematológicos ni ováricos.[85]
Los agonistas de la hormona liberadora de gonadotropina se usan para la supresión ovárica, pero se consideran experimentales y no se ha comprobado su eficacia.[86] En varios ensayos aleatorizados y metanálisis se exploraron los beneficios de los análogos de la hormona liberadora de gonadotropina durante la quimioterapia. No obstante, el uso de dichos análogos para la protección ovárica durante la quimioterapia todavía es controvertido. En 2 ensayos controlados aleatorizados se demostró que el funcionamiento menstrual, la ovulación y el embarazo fueron más probables en las pacientes con cáncer de mama que recibieron agonistas de la hormona liberadora de gonadotropina durante la quimioterapia, en comparación con aquellas que no recibieron este tratamiento; no obstante, no se conocen los beneficios para los desenlaces de fertilidad. Los estudios han sido limitados por un seguimiento inapropiado y la evaluación de mediciones sustitutas de fertilidad en lugar de tasas de embarazo. Si bien la Administración de Alimentos y Medicamentos de los Estados Unidos no ha aprobado el uso de los análogos de la hormona liberadora de gonadotropina para conservar la capacidad reproductiva, este tipo de medicamentos se usan de para esta indicación no autorizada.[87]
Riesgo de complicaciones en el embarazo
En numerosas investigaciones se evaluaron la prevalencia y los factores de riesgo de las complicaciones del embarazo en sobrevivientes adultas tratadas por cáncer infantil que conservaron su capacidad reproductiva. Se observaron complicaciones del embarazo, como hipertensión, mala posición del feto, muerte fetal o aborto espontáneo, trabajo de parto prematuro y bajo peso al nacer en relación con grupos específicos de diagnóstico y tratamiento.[88-93]
Evidencia (exceso de riesgo de complicaciones en el embarazo):
En un estudio de 4029 embarazos de 1915 mujeres sometidas a control en el marco del CCSS, los desenlaces fueron los siguientes: un 63 % de recién nacidos vivos, un 1 % de mortinatos, un 15 % de abortos espontáneos, un 17 % de abortos provocados y un 3 % de estado desconocido o en gestación.[88]
El riesgo de aborto espontáneo fue 3,6 veces más alto en las mujeres tratadas con irradiación craneoespinal y 1,7 veces más alto en aquellas tratadas con irradiación pélvica. La exposición a la quimioterapia sola no aumentó por sí sola el riesgo de aborto espontáneo.
Fue menos probable que las sobrevivientes tuvieran recién nacidos vivos, más probable que se sometieran a abortos médicos y más probable que tuvieran bebés de bajo peso al nacer que las hermanas.
La alteración del funcionamiento normal del útero después de la radioterapia u otros tratamientos que producen disminución del volumen uterino y deterioro del flujo sanguíneo en el útero parece ser el proceso fisiopatológico subyacente de muchas de estas complicaciones obstétricas.[94,95]
En el marco del National Wilms Tumor Study se obtuvieron informes de 1021 embarazos de más de 20 semanas de gestación. En este grupo, hubo 955 recién nacidos vivos de embarazos únicos.[96]
La hipertensión que complicó el embarazo, el trabajo de parto prematuro o la amenaza de parto prematuro, la mala posición del feto, el peso bajo al nacer (<2500 g) y el parto prematuro (<36 semanas) fueron más frecuentes en las mujeres que recibieron irradiación dirigida al flanco, en una forma dependiente de la dosis.
En otro estudio del CCSS se evaluaron los desenlaces de embarazos de compañeras de varones sobrevivientes.[89]
De 4106 hombres sexualmente activos, 1227 notificaron que habían engendrado 2323 embarazos que resultaron en un 69 % de recién nacidos vivos, un 13 % de abortos espontáneos, un 13 % de abortos provocados y un 5 % de estado desconocido o en gestación en el momento del análisis.
En comparación con las compañeras de los hermanos varones, hubo una disminución de la incidencia de recién nacidos vivos (RR, 0,77), pero no hubo diferencias significativas en el desenlace del embarazo según el tratamiento.
Los resultados de un estudio danés confirman la relación entre la irradiación uterina con el aborto espontáneo, pero no con otros tipos de aborto. En el Danish Cancer Registry, se evaluaron más de 34 000 embarazos, incluidos 1479 embarazos en 1688 mujeres sobrevivientes de cáncer infantil. Se identificaron los desenlaces de los embarazos de las sobrevivientes, de 2737 hermanas y de 16 700 mujeres de la población con fines de comparación.[90]
No se observaron diferencias significativas entre las sobrevivientes y las mujeres del grupo de comparación en la proporción de recién nacidos vivos, mortinatos o todos los tipos de aborto combinados.
Las sobrevivientes con antecedentes de radioterapia neuroendocrina o abdominal tuvieron un aumento del riesgo de aborto espontáneo.
Los desenlaces de los embarazos de las sobrevivientes fueron similares a los de las mujeres en el grupo de comparación, con excepción del aborto espontáneo.
En un análisis retrospectivo de una cohorte del CCSS de 1148 hombres y 1657 mujeres sobrevivientes de cáncer, se identificaron 4946 embarazos.[91]
La irradiación de los testículos en los hombres y la hipófisis en las mujeres, así como la quimioterapia con alquilantes no se relacionaron con un aumento de riesgo de mortinatos o muerte neonatal.
La irradiación uterina y ovárica en dosis superiores a 10 Gy aumentó de manera significativa el riesgo de muerte fetal y neonatal.
En las niñas tratadas antes de la menarquia, la irradiación del útero y los ovarios en dosis de tan solo 1 a 2,49 Gy aumentó de manera significativa el riesgo de mortinatos o muerte neonatal.
La mayoría de los embarazos notificados de sobrevivientes de TCMH y sus compañeros dieron como resultado recién nacidos vivos.[92]
En las mujeres sobrevivientes de TCMH expuestas a ICT, aumenta el riesgo de parto prematuro de lactantes con bajo peso al nacer.
Las sobrevivientes sometidas a TCMH tienen un aumento de riesgo de cesárea con indicación médica en comparación con la población normal (42 vs.16 %).
En un estudio observacional retrospectivo se incluyó a 178 mujeres sobrevivientes de leucemia infantil del programa francés de seguimiento a largo plazo. En el estudio se evaluaron mujeres que se habían sometido a TCMH prepuberal por leucemia aguda. Entre estas sobrevivientes, 116 habían recibido ICT y 62 habían recibido un protocolo de acondicionamiento a base de busulfano. La mediana de seguimiento desde el TCMH fue de 18 años.[62]
En esta cohorte, 22 mujeres (12 %) tuvieron, al menos, un embarazo espontáneo, con un total de 37 embarazos, entre los que se incluyen 17 recién nacidos vivos, 14 abortos espontáneos, 4 abortos legales y 2 abortos terapéuticos.
Las mujeres que no habían presentado insuficiencia ovárica prematura tenían más probabilidades de embarazo espontáneo que las mujeres que habían sufrido insuficiencia ovárica prematura inmediata o secundaria.
Es posible que la capacidad reproductiva se preserve y se logren embarazos fructíferos después de un TCMH, aunque los regímenes de acondicionamiento que incluyen ICT, ciclofosfamida y busulfano son muy gonadotóxicos. En un estudio se evaluaron los desenlaces de embarazos en un grupo de mujeres tratadas con TCMH.[97]
Entre 708 mujeres pospúberes en el momento del trasplante, 116 recuperaron el funcionamiento ovárico normal y 32 quedaron embarazadas.
Entre 82 mujeres prepúberes en el momento del trasplante, 23 tenían un funcionamiento ovárico normal y 9 quedaron embarazadas.
De los 72 embarazos de estas 41 mujeres, 16 se produjeron en aquellas tratadas con ICT y el 50 % resultaron en una interrupción temprana del embarazo.
De los 56 embarazos de mujeres tratadas con ciclofosfamida, sin ICT ni busulfano, el 21 % resultaron en interrupción temprana.
No hubo embarazos en las 73 mujeres tratadas con busulfano y ciclofosfamida, y solo una conservó el funcionamiento ovárico.
En un estudio del German Pediatric Oncology Group se demostró que la tasa de embarazos de mujeres sobrevivientes de linfoma de Hodgkin fue similar a la tasa de embarazos de la población general; sin embargo, fue más baja en las sobrevivientes sometidas a radioterapia pélvica.[98]
Los investigadores del CCSS británico evaluaron las complicaciones del embarazo y parto en mujeres sobrevivientes de cáncer infantil tratadas con radiación abdominal; en el estudio se vincularon los datos de la cohorte del CCSS británico con un registro nacional de hospitales.[99]
Las sobrevivientes tratadas con radiación abdominal tuvieron un riesgo significativamente más alto (RR, 2,1) de complicaciones del embarazo que las sobrevivientes que no recibieron esta radiación.
Los riesgos fueron elevados para la hipertensión, que complicó el embarazo de las sobrevivientes del tumor de Wilms (RR, 3,29) tratadas con radiación abdominal. También fueron elevados los riesgos de diabetes mellitus de la gestación (RR, 3,35) y la anemia (RR, 2,10) en todas las sobrevivientes tratadas con radiación abdominal.
En una revisión sistemática se compararon los datos de desenlaces publicados sobre embarazo y salud infantil para sobrevivientes de leucemia y linfoma durante la edad pediátrica o adultez temprana con los desenlaces de controles que no tenían antecedentes de cáncer.[100]
No se observaron riesgos más altos de aborto espontáneo, diabetes y anemia maternas, mortinatos, anomalías congénitas o cánceres infantiles en los descendientes de los sobrevivientes, en comparación con los controles.
Las tasas de recién nacidos vivos fueron más bajas, aunque el riesgo de parto prematuro y bajo peso al nacer fueron un poco más altos en los sobrevivientes que en los controles.
En un estudio se revisaron los efectos de la radioterapia, dirigida a una parte o todo el abdomen, en el embarazo y los resultados del embarazo en pacientes con antecedentes de tumor de Wilms. Se produjeron 1021 embarazos (con una duración de 20 semanas o más), incluidos 955 nacidos vivos; se evaluaron 700 series de historiales médicos de madres e hijos.[96]
Las mujeres sobrevivientes de un tumor de Wilms que recibieron radioterapia tienen un mayor riesgo de presentar hipertensión que complique el embarazo, parto prematuro o amenaza de parto y posición fetal anómala.
Los hijos de estas pacientes tenían más probabilidad de ser prematuros y tener peso bajo al nacer.
Mujeres adolescentes y adultas jóvenes sobrevivientes de cánceres
El mayor estudio para investigar de forma exhaustiva las tasas de natalidad y las complicaciones obstétricas en mujeres adolescentes y adultas jóvenes (AAJ) sobrevivientes de cáncer e informar de un amplio espectro de resultados obstétricos para tipos específicos de cáncer se llevó a cabo mediante el Teenage Young Adult Cancer Survivor Study (TYACSS). En el estudio se incluyó a más de 200 945 sobrevivientes de cáncer durante 5 años, diagnosticadas entre los 15 y los 39 años de edad, de Inglaterra y Gales. Los investigadores cuantificaron las deficiencias en las tasas de natalidad y los riesgos de complicaciones obstétricas de las mujeres sobrevivientes (n = 13 886) de 17 tipos específicos de cáncer en AAJ.[93]
Las sobrevivientes tuvieron el 68 % de los nacimientos esperados en la población general. Las sobrevivientes de otros cánceres genitourinarios (distintos del de cuello uterino, ovario, vejiga o riñón), cáncer de cuello uterino y cáncer de mama tuvieron menos del 50 % de los nacimientos esperados. Las sobrevivientes de leucemia tuvieron el 53 % de los nacimientos esperados.
Las sobrevivientes de cáncer de cuello uterino tuvieron riesgo de sufrir una serie de complicaciones obstétricas, como mala presentación del feto, parto obstruido, trastornos del líquido amniótico y de las membranas, rotura prematura de membranas, parto prematuro, trastornos placentarios como placenta previa y hemorragia preparto.
Las sobrevivientes de leucemia tenían riesgo de parto prematuro, parto obstruido, hemorragia posparto y retención de placenta.
Las sobrevivientes de todos los demás cánceres específicos no tuvieron más de 2 complicaciones obstétricas que superaran una proporción observada o esperada de 1,25 o superior.
A partir de estos datos, los investigadores indicaron que las sobrevivientes de cáncer de cuello uterino y leucemia deberían considerarse de riesgo alto de complicaciones obstétricas, mientras que las sobrevivientes de otros tipos de cáncer en AAJ deberían estar tranquilas respecto a este riesgo.
Descendencia de los sobrevivientes de cáncer infantil
En los sobrevivientes de cáncer infantil que logran tener descendencia, existe la preocupación de la ocurrencia de anomalías congénitas, enfermedades genéticas o riesgo de cáncer en los hijos de ambos sexos. Los descendientes de ambos sexos de los sobrevivientes de cáncer no tienen un aumento significativo de riesgo de anomalías congénitas a raíz de la exposición de sus progenitores a los tratamientos oncológicos mutagénicos.
Evidencia (hijos e hijas de sobrevivientes de cáncer que no tienen un aumento significativo de anomalías congénitas):
En un análisis retrospectivo de cohortes de casos verificados de anomalías congénitas en 4699 descendientes de ambos sexos de 1128 varones y 1627 mujeres que participaron en el CCSS se observaron los siguientes resultados:[101]
No se observaron relaciones significativas entre la radioterapia gonadal o la acumulación de exposición a alquilantes y las anomalías congénitas de los descendientes.
En un estudio se compararon 2198 descendientes de ambos sexos de sobrevivientes adultos tratados por cáncer infantil entre 1945 y 1975 con 4544 descendientes de ambos sexos de hermanos y hermanas que se usaron como controles.[102]
No hubo diferencias en la proporción de descendientes con síndromes citogenéticos, defectos en un solo gen o malformaciones simples.
De manera semejante, no hubo ningún efecto del tipo de tratamiento del cáncer infantil en la presentación de enfermedades genéticas en a descendencia.
En un estudio poblacional se comparó a 2630 hijos de ambos sexos nacidos vivos de sobrevivientes de cáncer infantil con 5504 hijos de ambos sexos nacidos vivos de hermanos y hermanas de sobrevivientes;[103]
No se encontraron diferencias en la proporción de cariotipos anómalos, la incidencia del síndrome de Down o el síndrome de Turner en descendientes de sobrevivientes y sus hermanos de ambos sexos.
En la misma cohorte poblacional, los sobrevivientes tratados con radioterapia abdominal o alquilantes no tuvieron un aumento del riesgo de tener descendientes con enfermedades genéticas, en comparación con los sobrevivientes no expuestos a estos fármacos.
En un estudio se evaluó a 5847 descendientes de sobrevivientes de cánceres infantiles tratados en 5 países escandinavos.[104]
En ausencia de un síndrome de cáncer hereditario (como un retinoblastoma hereditario), no se encontró un aumento de riesgo de contraer cáncer en la descendencia.
En comparación con la descendencia de los hermanos de ambos sexos, no hubo exceso de riesgo de trastornos de un solo gen, malformaciones congénitas ni síndromes cromosómicos en la descendencia de pacientes anteriores.[105]
En un estudio del European Group for Blood and Marrow Transplantation, se evaluaron los desenlaces del embarazo en 19 412 pacientes sometidos a trasplante alogénico y en 17 950 pacientes sometidos a trasplante autógeno.[92]
Los investigadores no observaron ningún aumento del riesgo de defectos congénitos, retraso del desarrollo ni cáncer entre la descendencia de receptores de ambos sexos de un TCMH.
En un estudio nacional de un registro de la población finlandesa, se comparó el riesgo de anomalías congénitas en la descendencia de 6862 sobrevivientes a largo plazo de canceres diagnosticados durante la niñez, la adolescencia y la adultez temprana tratados entre 1953 y 2004 con el riesgo de anomalías congénitas en la descendencia de 35 690 hermanos y hermanas.[106]
No se encontró ningún exceso de riesgo significativo de anomalías congénitas entre sobrevivientes de cáncer en la niñez o adolescencia (razón de prevalencia [RP], 1,17; IC 95 %, 0,92–1,49) y los sobrevivientes de cáncer en la adultez joven (RP, 1,17; IC 95 %, 0,83–1,23), en comparación con los hermanos y hermanas.
Hubo una relación entre el cáncer de los progenitores y las anomalías congénitas en los descendientes de los sobrevivientes diagnosticados en las décadas anteriores (1955-1964: RP, 2,77; IC 95 %, 1,26–6,11; y 1965–1974: RP, 1,55; IC 95 %, 0,94–2,56).
En el Cuadro 17 se resumen los efectos tardíos reproductivos y los exámenes médicos de detección correspondientes.
Disfunción hormonal testicular: Deficiencia o insuficiencia de testosterona, demora o interrupción de la pubertad
Estadio de Tanner
Testosterona matutina
LH
Alteración de la espermatogénesis Reducción de la fertilidad, oligospermia, azoospermia, esterilidad
Análisis del semen
FSH
Inhibina B
Deficiencias hormonales ováricas: Demora o interrupción de la pubertad; insuficiencia ovárica prematura o menopausia prematura. Reducción de la reserva folicular ovárica: Disminución de la reserva ovárica, esterilidad.
van Dorp W, Mulder RL, Kremer LC, et al.: Recommendations for Premature Ovarian Insufficiency Surveillance for Female Survivors of Childhood, Adolescent, and Young Adult Cancer: A Report From the International Late Effects of Childhood Cancer Guideline Harmonization Group in Collaboration With the PanCareSurFup Consortium. J Clin Oncol 34 (28): 3440-50, 2016. [PUBMED Abstract]
Skinner R, Mulder RL, Kremer LC, et al.: Recommendations for gonadotoxicity surveillance in male childhood, adolescent, and young adult cancer survivors: a report from the International Late Effects of Childhood Cancer Guideline Harmonization Group in collaboration with the PanCareSurFup Consortium. Lancet Oncol 18 (2): e75-e90, 2017. [PUBMED Abstract]
Green DM, Nolan VG, Goodman PJ, et al.: The cyclophosphamide equivalent dose as an approach for quantifying alkylating agent exposure: a report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer 61 (1): 53-67, 2014. [PUBMED Abstract]
Merchant TE, Wu S, Onar-Thomas A, et al.: Hypogonadism After Treatment for Medulloblastoma: Results From the SJMB03 Trial of Risk-Adapted Radiation Therapy. Int J Radiat Oncol Biol Phys 116 (3): 569-578, 2023. [PUBMED Abstract]
Lehmann V, Chemaitilly W, Lu L, et al.: Gonadal Functioning and Perceptions of Infertility Risk Among Adult Survivors of Childhood Cancer: A Report From the St Jude Lifetime Cohort Study. J Clin Oncol 37 (11): 893-902, 2019. [PUBMED Abstract]
Arap MA, Vicentini FC, Cocuzza M, et al.: Late hormonal levels, semen parameters, and presence of antisperm antibodies in patients treated for testicular torsion. J Androl 28 (4): 528-32, 2007 Jul-Aug. [PUBMED Abstract]
Tryfonas G, Violaki A, Tsikopoulos G, et al.: Late postoperative results in males treated for testicular torsion during childhood. J Pediatr Surg 29 (4): 553-6, 1994. [PUBMED Abstract]
Narayan P, Lange PH, Fraley EE: Ejaculation and fertility after extended retroperitoneal lymph node dissection for testicular cancer. J Urol 127 (4): 685-8, 1982. [PUBMED Abstract]
Nijman JM, Jager S, Boer PW, et al.: The treatment of ejaculation disorders after retroperitoneal lymph node dissection. Cancer 50 (12): 2967-71, 1982. [PUBMED Abstract]
Fankhauser CD, Afferi L, Stroup SP, et al.: Minimally invasive retroperitoneal lymph node dissection for men with testis cancer: a retrospective cohort study of safety and feasibility. World J Urol 40 (6): 1505-1512, 2022. [PUBMED Abstract]
Schlegel PN, Walsh PC: Neuroanatomical approach to radical cystoprostatectomy with preservation of sexual function. J Urol 138 (6): 1402-6, 1987. [PUBMED Abstract]
Frees S, Rubenwolf P, Ziesel C, et al.: Erectile function after treatment for rhabdomyosarcoma of prostate and bladder. J Pediatr Urol 12 (6): 404.e1-404.e6, 2016. [PUBMED Abstract]
Hahn EW, Feingold SM, Simpson L, et al.: Recovery from aspermia induced by low-dose radiation in seminoma patients. Cancer 50 (2): 337-40, 1982. [PUBMED Abstract]
Blatt J, Sherins RJ, Niebrugge D, et al.: Leydig cell function in boys following treatment for testicular relapse of acute lymphoblastic leukemia. J Clin Oncol 3 (9): 1227-31, 1985. [PUBMED Abstract]
Izard MA: Leydig cell function and radiation: a review of the literature. Radiother Oncol 34 (1): 1-8, 1995. [PUBMED Abstract]
Romerius P, Ståhl O, Moëll C, et al.: High risk of azoospermia in men treated for childhood cancer. Int J Androl 34 (1): 69-76, 2011. [PUBMED Abstract]
Shafford EA, Kingston JE, Malpas JS, et al.: Testicular function following the treatment of Hodgkin's disease in childhood. Br J Cancer 68 (6): 1199-204, 1993. [PUBMED Abstract]
Whitehead E, Shalet SM, Jones PH, et al.: Gonadal function after combination chemotherapy for Hodgkin's disease in childhood. Arch Dis Child 57 (4): 287-91, 1982. [PUBMED Abstract]
Aubier F, Flamant F, Brauner R, et al.: Male gonadal function after chemotherapy for solid tumors in childhood. J Clin Oncol 7 (3): 304-9, 1989. [PUBMED Abstract]
Jaffe N, Sullivan MP, Ried H, et al.: Male reproductive function in long-term survivors of childhood cancer. Med Pediatr Oncol 16 (4): 241-7, 1988. [PUBMED Abstract]
Pryzant RM, Meistrich ML, Wilson G, et al.: Long-term reduction in sperm count after chemotherapy with and without radiation therapy for non-Hodgkin's lymphomas. J Clin Oncol 11 (2): 239-47, 1993. [PUBMED Abstract]
Meistrich ML, Wilson G, Brown BW, et al.: Impact of cyclophosphamide on long-term reduction in sperm count in men treated with combination chemotherapy for Ewing and soft tissue sarcomas. Cancer 70 (11): 2703-12, 1992. [PUBMED Abstract]
Kenney LB, Laufer MR, Grant FD, et al.: High risk of infertility and long term gonadal damage in males treated with high dose cyclophosphamide for sarcoma during childhood. Cancer 91 (3): 613-21, 2001. [PUBMED Abstract]
Garolla A, Pizzato C, Ferlin A, et al.: Progress in the development of childhood cancer therapy. Reprod Toxicol 22 (2): 126-32, 2006. [PUBMED Abstract]
Williams D, Crofton PM, Levitt G: Does ifosfamide affect gonadal function? Pediatr Blood Cancer 50 (2): 347-51, 2008. [PUBMED Abstract]
Jahnukainen K, Heikkinen R, Henriksson M, et al.: Semen quality and fertility in adult long-term survivors of childhood acute lymphoblastic leukemia. Fertil Steril 96 (4): 837-42, 2011. [PUBMED Abstract]
Green DM, Liu W, Kutteh WH, et al.: Cumulative alkylating agent exposure and semen parameters in adult survivors of childhood cancer: a report from the St Jude Lifetime Cohort Study. Lancet Oncol 15 (11): 1215-23, 2014. [PUBMED Abstract]
Sanders JE, Buckner CD, Leonard JM, et al.: Late effects on gonadal function of cyclophosphamide, total-body irradiation, and marrow transplantation. Transplantation 36 (3): 252-5, 1983. [PUBMED Abstract]
Viviani S, Santoro A, Ragni G, et al.: Gonadal toxicity after combination chemotherapy for Hodgkin's disease. Comparative results of MOPP vs ABVD. Eur J Cancer Clin Oncol 21 (5): 601-5, 1985. [PUBMED Abstract]
Mackie EJ, Radford M, Shalet SM: Gonadal function following chemotherapy for childhood Hodgkin's disease. Med Pediatr Oncol 27 (2): 74-8, 1996. [PUBMED Abstract]
Sherins RJ, Olweny CL, Ziegler JL: Gynecomastia and gonadal dysfunction in adolescent boys treated with combination chemotherapy for Hodgkin's disease. N Engl J Med 299 (1): 12-6, 1978. [PUBMED Abstract]
Dhabhar BN, Malhotra H, Joseph R, et al.: Gonadal function in prepubertal boys following treatment for Hodgkin's disease. Am J Pediatr Hematol Oncol 15 (3): 306-10, 1993. [PUBMED Abstract]
Heikens J, Behrendt H, Adriaanse R, et al.: Irreversible gonadal damage in male survivors of pediatric Hodgkin's disease. Cancer 78 (9): 2020-4, 1996. [PUBMED Abstract]
da Cunha MF, Meistrich ML, Fuller LM, et al.: Recovery of spermatogenesis after treatment for Hodgkin's disease: limiting dose of MOPP chemotherapy. J Clin Oncol 2 (6): 571-7, 1984. [PUBMED Abstract]
Brämswig JH, Heimes U, Heiermann E, et al.: The effects of different cumulative doses of chemotherapy on testicular function. Results in 75 patients treated for Hodgkin's disease during childhood or adolescence. Cancer 65 (6): 1298-302, 1990. [PUBMED Abstract]
Green DM, Zhu L, Wang M, et al.: Effect of cranial irradiation on sperm concentration of adult survivors of childhood acute lymphoblastic leukemia: a report from the St. Jude Lifetime Cohort Study†. Hum Reprod 32 (6): 1192-1201, 2017. [PUBMED Abstract]
Lopez R, Plat G, Bertrand Y, et al.: Testosterone deficiency in men surviving childhood acute leukemia after treatment with hematopoietic stem cell transplantation or testicular radiation: an L.E.A. study. Bone Marrow Transplant 56 (6): 1422-1425, 2021. [PUBMED Abstract]
Haavisto A, Lampic C, Wettergren L, et al.: Reproductive late effects and testosterone replacement therapy in male childhood cancer survivors: A population-based study (the Fex-Can study). Int J Cancer 154 (12): 2121-2131, 2024. [PUBMED Abstract]
Cattoni A, Nicolosi ML, Capitoli G, et al.: Pubertal attainment and Leydig cell function following pediatric hematopoietic stem cell transplantation: a three-decade longitudinal assessment. Front Endocrinol (Lausanne) 14: 1292683, 2023. [PUBMED Abstract]
Ishiguro H, Yasuda Y, Tomita Y, et al.: Gonadal shielding to irradiation is effective in protecting testicular growth and function in long-term survivors of bone marrow transplantation during childhood or adolescence. Bone Marrow Transplant 39 (8): 483-90, 2007. [PUBMED Abstract]
Panasiuk A, Nussey S, Veys P, et al.: Gonadal function and fertility after stem cell transplantation in childhood: comparison of a reduced intensity conditioning regimen containing melphalan with a myeloablative regimen containing busulfan. Br J Haematol 170 (5): 719-26, 2015. [PUBMED Abstract]
Green DM, Zhu L, Zhang N, et al.: Lack of specificity of plasma concentrations of inhibin B and follicle-stimulating hormone for identification of azoospermic survivors of childhood cancer: a report from the St Jude lifetime cohort study. J Clin Oncol 31 (10): 1324-8, 2013. [PUBMED Abstract]
van Dorp W, van der Geest IM, Laven JS, et al.: Gonadal function recovery in very long-term male survivors of childhood cancer. Eur J Cancer 49 (6): 1280-6, 2013. [PUBMED Abstract]
Chemaitilly W, Liu Q, van Iersel L, et al.: Leydig Cell Function in Male Survivors of Childhood Cancer: A Report From the St Jude Lifetime Cohort Study. J Clin Oncol 37 (32): 3018-3031, 2019. [PUBMED Abstract]
Park JY, Kim DY, Suh DS, et al.: Outcomes of pediatric and adolescent girls with malignant ovarian germ cell tumors. Gynecol Oncol 137 (3): 418-22, 2015. [PUBMED Abstract]
Wallace WH, Shalet SM, Crowne EC, et al.: Ovarian failure following abdominal irradiation in childhood: natural history and prognosis. Clin Oncol (R Coll Radiol) 1 (2): 75-9, 1989. [PUBMED Abstract]
Scott JE: Pubertal development in children treated for nephroblastoma. J Pediatr Surg 16 (2): 122-5, 1981. [PUBMED Abstract]
Hamre MR, Robison LL, Nesbit ME, et al.: Effects of radiation on ovarian function in long-term survivors of childhood acute lymphoblastic leukemia: a report from the Childrens Cancer Study Group. J Clin Oncol 5 (11): 1759-65, 1987. [PUBMED Abstract]
Wallace WH, Shalet SM, Tetlow LJ, et al.: Ovarian function following the treatment of childhood acute lymphoblastic leukaemia. Med Pediatr Oncol 21 (5): 333-9, 1993. [PUBMED Abstract]
Sklar CA, Mertens AC, Mitby P, et al.: Premature menopause in survivors of childhood cancer: a report from the childhood cancer survivor study. J Natl Cancer Inst 98 (13): 890-6, 2006. [PUBMED Abstract]
Chemaitilly W, Mertens AC, Mitby P, et al.: Acute ovarian failure in the childhood cancer survivor study. J Clin Endocrinol Metab 91 (5): 1723-8, 2006. [PUBMED Abstract]
Vatanen A, Wilhelmsson M, Borgström B, et al.: Ovarian function after allogeneic hematopoietic stem cell transplantation in childhood and adolescence. Eur J Endocrinol 170 (2): 211-8, 2014. [PUBMED Abstract]
Sanders JE, Buckner CD, Amos D, et al.: Ovarian function following marrow transplantation for aplastic anemia or leukemia. J Clin Oncol 6 (5): 813-8, 1988. [PUBMED Abstract]
Levine JM, Whitton JA, Ginsberg JP, et al.: Nonsurgical premature menopause and reproductive implications in survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 124 (5): 1044-1052, 2018. [PUBMED Abstract]
van den Berg MH, van Dijk M, Byrne J, et al.: Treatment-related fertility impairment in long-term female childhood, adolescent and young adult cancer survivors: investigating dose-effect relationships in a European case-control study (PanCareLIFE). Hum Reprod 36 (6): 1561-1573, 2021. [PUBMED Abstract]
van der Kaaij MA, Heutte N, Meijnders P, et al.: Premature ovarian failure and fertility in long-term survivors of Hodgkin's lymphoma: a European Organisation for Research and Treatment of Cancer Lymphoma Group and Groupe d'Etude des Lymphomes de l'Adulte Cohort Study. J Clin Oncol 30 (3): 291-9, 2012. [PUBMED Abstract]
Thomas-Teinturier C, El Fayech C, Oberlin O, et al.: Age at menopause and its influencing factors in a cohort of survivors of childhood cancer: earlier but rarely premature. Hum Reprod 28 (2): 488-95, 2013. [PUBMED Abstract]
Weil BR, Murphy AJ, Liu Q, et al.: Late Health Outcomes Among Survivors of Wilms Tumor Diagnosed Over Three Decades: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 41 (14): 2638-2650, 2023. [PUBMED Abstract]
Chemaitilly W, Li Z, Krasin MJ, et al.: Premature Ovarian Insufficiency in Childhood Cancer Survivors: A Report From the St. Jude Lifetime Cohort. J Clin Endocrinol Metab 102 (7): 2242-2250, 2017. [PUBMED Abstract]
Jimenez-Kurlander L, DeRosa A, Kostrzewa CE, et al.: Ovarian function in female survivors of high-risk neuroblastoma. Pediatr Blood Cancer 71 (9): e31181, 2024. [PUBMED Abstract]
Bresters D, Emons JA, Nuri N, et al.: Ovarian insufficiency and pubertal development after hematopoietic stem cell transplantation in childhood. Pediatr Blood Cancer 61 (11): 2048-53, 2014. [PUBMED Abstract]
Chabut M, Schneider P, Courbiere B, et al.: Ovarian Function and Spontaneous Pregnancy After Hematopoietic Stem Cell Transplantation for Leukemia Before Puberty: An L.E.A. Cohort Study. Transplant Cell Ther 29 (6): 378.e1-378.e9, 2023. [PUBMED Abstract]
Clark RA, Mostoufi-Moab S, Yasui Y, et al.: Predicting acute ovarian failure in female survivors of childhood cancer: a cohort study in the Childhood Cancer Survivor Study (CCSS) and the St Jude Lifetime Cohort (SJLIFE). Lancet Oncol 21 (3): 436-445, 2020. [PUBMED Abstract]
Im C, Lu Z, Mostoufi-Moab S, et al.: Development and validation of age-specific risk prediction models for primary ovarian insufficiency in long-term survivors of childhood cancer: a report from the Childhood Cancer Survivor Study and St Jude Lifetime Cohort. Lancet Oncol 24 (12): 1434-1442, 2023. [PUBMED Abstract]
van den Berg MH, Overbeek A, Lambalk CB, et al.: Long-term effects of childhood cancer treatment on hormonal and ultrasound markers of ovarian reserve. Hum Reprod 33 (8): 1474-1488, 2018. [PUBMED Abstract]
Chow EJ, Stratton KL, Leisenring WM, et al.: Pregnancy after chemotherapy in male and female survivors of childhood cancer treated between 1970 and 1999: a report from the Childhood Cancer Survivor Study cohort. Lancet Oncol 17 (5): 567-76, 2016. [PUBMED Abstract]
Armuand G, Skoog-Svanberg A, Bladh M, et al.: Reproductive Patterns Among Childhood and Adolescent Cancer Survivors in Sweden: A Population-Based Matched-Cohort Study. J Clin Oncol 35 (14): 1577-1583, 2017. [PUBMED Abstract]
Nies M, Cantineau AEP, Arts EGJM, et al.: Long-Term Effects of Radioiodine Treatment on Female Fertility in Survivors of Childhood Differentiated Thyroid Carcinoma. Thyroid 30 (8): 1169-1176, 2020. [PUBMED Abstract]
Critchley HO, Wallace WH, Shalet SM, et al.: Abdominal irradiation in childhood; the potential for pregnancy. Br J Obstet Gynaecol 99 (5): 392-4, 1992. [PUBMED Abstract]
Bjornard KL, Howell CR, Klosky JL, et al.: Psychosexual Functioning of Female Childhood Cancer Survivors: A Report From the St. Jude Lifetime Cohort Study. J Sex Med 17 (10): 1981-1994, 2020. [PUBMED Abstract]
Chevalier LL, McCormick K, Cooney TM, et al.: Sexual health in adult survivors of childhood cancer: A Project REACH study. Cancer 130 (17): 3023-3033, 2024. [PUBMED Abstract]
Loren AW, Mangu PB, Beck LN, et al.: Fertility preservation for patients with cancer: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol 31 (19): 2500-10, 2013. [PUBMED Abstract]
Mulder RL, Font-Gonzalez A, Hudson MM, et al.: Fertility preservation for female patients with childhood, adolescent, and young adult cancer: recommendations from the PanCareLIFE Consortium and the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol 22 (2): e45-e56, 2021. [PUBMED Abstract]
Mulder RL, Font-Gonzalez A, Green DM, et al.: Fertility preservation for male patients with childhood, adolescent, and young adult cancer: recommendations from the PanCareLIFE Consortium and the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol 22 (2): e57-e67, 2021. [PUBMED Abstract]
Mulder RL, Font-Gonzalez A, van Dulmen-den Broeder E, et al.: Communication and ethical considerations for fertility preservation for patients with childhood, adolescent, and young adult cancer: recommendations from the PanCareLIFE Consortium and the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol 22 (2): e68-e80, 2021. [PUBMED Abstract]
Agarwa A: Semen banking in patients with cancer: 20-year experience. Int J Androl 23 (Suppl 2): 16-9, 2000. [PUBMED Abstract]
Müller J, Sønksen J, Sommer P, et al.: Cryopreservation of semen from pubertal boys with cancer. Med Pediatr Oncol 34 (3): 191-4, 2000. [PUBMED Abstract]
Dar S, Orvieto R, Levron J, et al.: IVF outcome in azoospermic cancer survivors. Eur J Obstet Gynecol Reprod Biol 220: 84-87, 2018. [PUBMED Abstract]
Borgmann-Staudt A, Michael S, Sommerhaeuser G, et al.: The Use of Assisted Reproductive Technology by European Childhood Cancer Survivors. Curr Oncol 29 (8): 5748-5762, 2022. [PUBMED Abstract]
Gunnes MW, Lie RT, Bjørge T, et al.: Reproduction and marriage among male survivors of cancer in childhood, adolescence and young adulthood: a national cohort study. Br J Cancer 114 (3): 348-56, 2016. [PUBMED Abstract]
Kitlinski M, Giwercman A, Elenkov A: Paternity through use of assisted reproduction technology in male adult and childhood cancer survivors: a nationwide register study. Hum Reprod 38 (5): 973-981, 2023. [PUBMED Abstract]
Domingo J, Ayllón Y, Domingo S, et al.: New approaches to female fertility preservation. Clin Transl Oncol 11 (3): 154-9, 2009. [PUBMED Abstract]
Meirow D, Ra'anani H, Shapira M, et al.: Transplantations of frozen-thawed ovarian tissue demonstrate high reproductive performance and the need to revise restrictive criteria. Fertil Steril 106 (2): 467-74, 2016. [PUBMED Abstract]
Pacheco F, Oktay K: Current Success and Efficiency of Autologous Ovarian Transplantation: A Meta-Analysis. Reprod Sci 24 (8): 1111-1120, 2017. [PUBMED Abstract]
Oktay K, Karlikaya G: Ovarian function after transplantation of frozen, banked autologous ovarian tissue. N Engl J Med 342 (25): 1919, 2000. [PUBMED Abstract]
Demeestere I, Brice P, Peccatori FA, et al.: No Evidence for the Benefit of Gonadotropin-Releasing Hormone Agonist in Preserving Ovarian Function and Fertility in Lymphoma Survivors Treated With Chemotherapy: Final Long-Term Report of a Prospective Randomized Trial. J Clin Oncol 34 (22): 2568-74, 2016. [PUBMED Abstract]
Practice Committee of the American Society for Reproductive Medicine. Electronic address: asrm@asrm.org: Fertility preservation in patients undergoing gonadotoxic therapy or gonadectomy: a committee opinion. Fertil Steril 112 (6): 1022-1033, 2019. [PUBMED Abstract]
Green DM, Kawashima T, Stovall M, et al.: Fertility of female survivors of childhood cancer: a report from the childhood cancer survivor study. J Clin Oncol 27 (16): 2677-85, 2009. [PUBMED Abstract]
Green DM, Kawashima T, Stovall M, et al.: Fertility of male survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. J Clin Oncol 28 (2): 332-9, 2010. [PUBMED Abstract]
Winther JF, Boice JD, Svendsen AL, et al.: Spontaneous abortion in a Danish population-based cohort of childhood cancer survivors. J Clin Oncol 26 (26): 4340-6, 2008. [PUBMED Abstract]
Signorello LB, Mulvihill JJ, Green DM, et al.: Stillbirth and neonatal death in relation to radiation exposure before conception: a retrospective cohort study. Lancet 376 (9741): 624-30, 2010. [PUBMED Abstract]
Salooja N, Szydlo RM, Socie G, et al.: Pregnancy outcomes after peripheral blood or bone marrow transplantation: a retrospective survey. Lancet 358 (9278): 271-6, 2001. [PUBMED Abstract]
Sunguc C, Winter DL, Heymer EJ, et al.: Risks of adverse obstetric outcomes among female survivors of adolescent and young adult cancer in England (TYACSS): a population-based, retrospective cohort study. Lancet Oncol 25 (8): 1080-1091, 2024. [PUBMED Abstract]
Beneventi F, Locatelli E, Giorgiani G, et al.: Adolescent and adult uterine volume and uterine artery Doppler blood flow among subjects treated with bone marrow transplantation or chemotherapy in pediatric age: a case-control study. Fertil Steril 103 (2): 455-61, 2015. [PUBMED Abstract]
van de Loo LEXM, van den Berg MH, Overbeek A, et al.: Uterine function, pregnancy complications, and pregnancy outcomes among female childhood cancer survivors. Fertil Steril 111 (2): 372-380, 2019. [PUBMED Abstract]
Green DM, Lange JM, Peabody EM, et al.: Pregnancy outcome after treatment for Wilms tumor: a report from the national Wilms tumor long-term follow-up study. J Clin Oncol 28 (17): 2824-30, 2010. [PUBMED Abstract]
Sanders JE, Hawley J, Levy W, et al.: Pregnancies following high-dose cyclophosphamide with or without high-dose busulfan or total-body irradiation and bone marrow transplantation. Blood 87 (7): 3045-52, 1996. [PUBMED Abstract]
Brämswig JH, Riepenhausen M, Schellong G: Parenthood in adult female survivors treated for Hodgkin's lymphoma during childhood and adolescence: a prospective, longitudinal study. Lancet Oncol 16 (6): 667-75, 2015. [PUBMED Abstract]
Reulen RC, Bright CJ, Winter DL, et al.: Pregnancy and Labor Complications in Female Survivors of Childhood Cancer: The British Childhood Cancer Survivor Study. J Natl Cancer Inst 109 (11): , 2017. [PUBMED Abstract]
Shliakhtsitsava K, Romero SAD, Dewald SR, et al.: Pregnancy and child health outcomes in pediatric and young adult leukemia and lymphoma survivors: a systematic review. Leuk Lymphoma 59 (2): 381-397, 2018. [PUBMED Abstract]
Signorello LB, Mulvihill JJ, Green DM, et al.: Congenital anomalies in the children of cancer survivors: a report from the childhood cancer survivor study. J Clin Oncol 30 (3): 239-45, 2012. [PUBMED Abstract]
Winther JF, Boice JD, Mulvihill JJ, et al.: Chromosomal abnormalities among offspring of childhood-cancer survivors in Denmark: a population-based study. Am J Hum Genet 74 (6): 1282-5, 2004. [PUBMED Abstract]
Winther JF, Olsen JH, Wu H, et al.: Genetic disease in the children of Danish survivors of childhood and adolescent cancer. J Clin Oncol 30 (1): 27-33, 2012. [PUBMED Abstract]
Sankila R, Olsen JH, Anderson H, et al.: Risk of cancer among offspring of childhood-cancer survivors. Association of the Nordic Cancer Registries and the Nordic Society of Paediatric Haematology and Oncology. N Engl J Med 338 (19): 1339-44, 1998. [PUBMED Abstract]
Byrne J, Rasmussen SA, Steinhorn SC, et al.: Genetic disease in offspring of long-term survivors of childhood and adolescent cancer. Am J Hum Genet 62 (1): 45-52, 1998. [PUBMED Abstract]
Seppänen VI, Artama MS, Malila NK, et al.: Risk for congenital anomalies in offspring of childhood, adolescent and young adult cancer survivors. Int J Cancer 139 (8): 1721-30, 2016. [PUBMED Abstract]
Efectos tardíos en el aparato respiratorio
El funcionamiento respiratorio podría verse comprometido en los sobrevivientes a largo plazo de cáncer infantil que recibieron los siguientes tratamientos:
Ciertos fármacos quimioterapéuticos.
Radioterapia torácica.
Cirugía pulmonar o de la pared torácica.
Trasplante de células madre hematopoyéticas (TCMH).
Los efectos de una lesión pulmonar temprana por el tratamiento del cáncer tal vez se exacerben por el deterioro del funcionamiento pulmonar relacionado con el envejecimiento normal, otras afecciones crónicas concomitantes o el consumo de tabaco. La calidad de la evidencia científica actual relacionada con este resultado está limitada por la recopilación de datos retrospectiva, las muestras pequeñas, el sesgo de selección de la cohorte y el sesgo de participación, la descripción de resultados tras abordajes de tratamiento anticuados y la variabilidad en el tiempo desde el tratamiento y el método de comprobación. No se han realizado estudios de cohortes grandes que incluyan evaluaciones clínicas acompañadas de evaluaciones funcionales y de calidad de vida.
La verdadera prevalencia o incidencia de la disfunción pulmonar en los sobrevivientes de cáncer diagnosticado durante la niñez y adolescencia no es clara. En los niños tratados con TCMH, se observa una enfermedad clínica significativa. En estudios poblacionales se demostró que los sobrevivientes presentaron exceso de morbilidad y mortalidad por afecciones respiratorias.[1,2]
En un estudio en el que se vincularon las notificaciones a un registro poblacional de cáncer y las hospitalizaciones de 4235 sobrevivientes a 5 años de cáncer infantil (edad de 0–29 años) diagnosticados entre 1990 y 2011, se compararon las tasas de hospitalización de los sobrevivientes y los controles poblacionales ajustados por edad y sexo.[1]
La incidencia acumulada de cualquier tipo de afección respiratoria fue del 49 % a los 40 años entre los sobrevivientes, que fue 1,86 veces (intervalo de confianza [IC] 95 %, 1,73–2,01) más alta que en los controles.
El tratamiento con quimioterapia pulmonar tóxica aumentó el riesgo de hospitalización por cualquier afección respiratoria (cociente de riesgos instantáneos de subdistribución [CRIs], 1,26; IC 95 %, 1,03–1,53) y neumonía (CRIs, 1,48; IC 95 %, 1,01–2,17).
La mortalidad posterior fue más alta en los pacientes hospitalizados por neumonía en comparación con otras afecciones respiratorias (28 vs. 15 %, respectivamente).
En otro estudio poblacional se investigó la mortalidad respiratoria a largo plazo antes de los 40 años entre sobrevivientes a 5 años de cáncer que participaron en el British Childhood Cancer Survivor Study (BCCSS) y el Teenage and Young Adult Cancer Survivor Study (TYACSS).[2]
Se observó exceso de muertes debidas a causas respiratorias en los sobrevivientes del BCCSS (razón de mortalidad estandarizada [RME], 6,8; IC 95 %, 5,8–7,9) y TYACSS (RME, 1,7; IC 95 %, 1,6–1,8), con riesgos que oscilaron según el tipo de causa de muerte respiratoria (por ejemplo, neumonía, neumonitis, fibrosis y enfermedad respiratoria inferior crónica).
Los sobrevivientes de tumores del SNC del estudio BCCSS y los sobrevivientes de cáncer de pulmón, leucemia y cánceres de cabeza y cuello en el estudio TYACSS presentaron el mayor número de muertes debidas a causas respiratorias.
Se observó aumento en el exceso de muertes debidas a causas respiratorias con la edad avanzada, pero también fue tranquilizador que se presentara una reducción en el exceso de muertes en las cohortes de sobrevivientes con diagnóstico más reciente.
Evidencia (estudios seleccionados de cohortes que describen desenlaces a largo plazo del funcionamiento pulmonar):
En un estudio longitudinal en el que se evaluó la magnitud y la trayectoria de la disfunción pulmonar en 121 sobrevivientes de cáncer en la niñez (mediana de tiempo desde el diagnóstico hasta la última evaluación, 17,1 años) que recibieron un tratamiento potencialmente tóxico para los pulmones (por ejemplo, bleomicina, busulfano y radioterapia pulmonar), se encontró que los sobrevivientes eran significativamente más propensos a presentar defectos restrictivos o en la difusión que los controles sanos.[3]
La edad menor de 16 años en el momento del diagnóstico y la exposición a más de 20 Gy de radiación dirigida al tórax fueron los factores relacionados con aumento de la probabilidad de defectos restrictivos; mientras que el sexo femenino y la dosis de radiación dirigida al tórax se relacionaron con las alteraciones en la difusión.
El deterioro del funcionamiento pulmonar a largo plazo se relacionó en gran medida con los cambios en la capacidad de difusión. Se observó que, con el tiempo, la probabilidad de deterioro en la función de difusión aumentó 4 veces en mujeres y 24 veces en sobrevivientes tratados con más de 20 Gy de radiación dirigida al tórax. En comparación con los sobrevivientes con difusión normal, aquellos con defectos en la difusión también fueron significativamente más propensos a ser sintomáticos y tener puntuaciones más precarias de la calidad de vida relacionada con la salud, con disminuciones en los dominios de funcionamiento físico, limitación de sus funciones sociales como consecuencia de la salud física, así como menor energía o aumento de la fatiga.
Los investigadores del Childhood Cancer Survivor Study (CCSS) compararon desenlaces pulmonares autonotificados y su efecto en las actividades de la vida diaria en sobrevivientes de cáncer a 5 años (n = 14 316; mediana desde el diagnóstico, 25 años) y una cohorte de hermanos (n = 4027).[4]
Fue más probable que los sobrevivientes notificaran tos crónica, necesidad de oxígeno, fibrosis pulmonar y neumonía recidivante que los hermanos de ambos sexos a pesar de las tasas más bajas de consumo de tabaco.
A los 45 años, la incidencia acumulada de cualquier afección pulmonar fue del 29,6 % para los sobrevivientes. Fue más probable que los sobrevivientes con afecciones pulmonares crónicas (por ejemplo, tos crónica) notificaran limitaciones en sus actividades que aquellos sin esas afecciones.
Las complicaciones pulmonares contribuyeron a un exceso de riesgo de muerte de seis veces para los sobrevivientes y demostraron relaciones significativas con la exposición a derivados del platino y la radiación pulmonar.
Los investigadores del estudio de cohorte St. Jude Life (SJLIFE) evaluaron los factores de riesgo de deficiencias de funcionamiento de los pulmones comprobadas clínicamente y el efecto funcional del deterioro pulmonar en 606 adultos sobrevivientes de cáncer infantil (mediana de edad, 34 años en el momento de la evaluación; mediana de tiempo desde el diagnóstico de cáncer, 21,9 años).[5]
Las deficiencias de funcionamiento pulmonar se identificaron mediante las siguientes evaluaciones:
Volumen espiratorio máximo en el primer segundo (VEF1) de menos del 80 % de lo predicho (50,7 % de los pacientes).
Capacidad vital forzada (CVF) de menos del 80 % de lo predicho (47,2 % de los pacientes).
Prueba por respiración única de la capacidad de difusión pulmonar para el monóxido de carbono (CDCO) ajustada para la hemoglobina de menos del 75 % de lo predicho (44,6 % de los pacientes).
Se encontraron anomalías pulmonares obstructivas (VEF1/CVF <0,7) en el 0,8 % de los pacientes y anomalías restrictivas (capacidad pulmonar total [CPT] <75 %) en el 31,2 % de los pacientes.
Los factores de riesgo para las anomalías en el funcionamiento de los pulmones incluyen los siguientes:
El porcentaje estimado de tejido pulmonar que recibió al menos 10 Gy de radioterapia (V10) y el tiempo transcurrido desde el diagnóstico para el VEF1.
Edad en el momento del diagnóstico y V10 para la CPT.
El aumento del índice de masa corporal, V10 y el tiempo transcurrido desde el diagnóstico para la CDCO corregida por hemoglobina.
Las anomalías en las pruebas de funcionamiento pulmonar se relacionaron con una disminución de 6 minutos en la distancia de caminata.
Complicaciones respiratorias después de la radioterapia
Es posible que la radioterapia que expone el parénquima pulmonar a la radiación produzca disfunción pulmonar relacionada con la reducción del volumen pulmonar, deterioro de la adaptabilidad dinámica y deformidad, tanto del pulmón como de la pared torácica.
La posibilidad de secuelas pulmonares crónicas se relaciona con la dosis de radiación administrada, el volumen de pulmón irradiado y las dosis de radioterapia fraccionada.[6]
El tratamiento de modalidad combinada, que incluye radioterapia y quimioterapia pulmonar tóxica o cirugía en la pared del tórax o en el tórax, aumenta el riesgo de deterioro del funcionamiento pulmonar.[7,8]
Entre las complicaciones pulmonares notificadas después del tratamiento de neoplasias malignas infantiles, se incluyen la enfermedad pulmonar restrictiva u obstructiva crónica, la fibrosis pulmonar y el neumotórax espontáneo.[9]
Estas secuelas no son comunes después de los tratamientos contemporáneos y aparecen con más frecuencia como lesiones subclínicas que se detectan solo mediante pruebas de imágenes o pruebas formales para analizar el funcionamiento pulmonar.
El grupo de trabajo pulmonar Pediatric Normal Tissue Effects in the Clinic (PENTEC) revisó factores dosimétricos y clínicos para crear modelos predictivos de la toxicidad pulmonar relacionada con la radioterapia en niños.[10]
Tras la radiación pulmonar parcial, la toxicidad pulmonar aguda fue infrecuente, con una incidencia de neumonitis por radiación de grado 2 a 3 inferior al 1 %.
Las toxicidades tardías, incluso el deterioro subclínico o asintomático de la función pulmonar, fueron más frecuentes (<4 %) y su incidencia y gravedad aumentaron con el tiempo.
El riesgo de complicaciones pulmonares fue bajo entre los niños tratados con una dosis pulmonar media inferior a 14 Gy y un V20 Gy pulmonar total inferior al 30 %.
Dentro del espectro de la dosis que se analizó, la toxicidad pulmonar fue mayor tras la irradiación dirigida a todo el pulmón, pero no se identificó ninguna relación dosis-respuesta.
La bleomicina y otros fármacos quimioterapéuticos en los regímenes de dosis actuales no contribuyeron de forma sustancial a los resultados pulmonares adversos tras la irradiación pulmonar parcial. Sin embargo, este riesgo aumentó con la irradiación a todo el pulmón.
Evidencia (estudios seleccionados de cohortes que describen desenlaces pulmonares):
En los sobrevivientes de linfoma de Hodgkin infantil, se notifica que la prevalencia de síntomas pulmonares es baja cuando se usan técnicas contemporáneas de radiación dirigida al campo afectado. Sin embargo, algunos pacientes exhibieron disfunción subclínica importante.[11]
Estos hallazgos se confirmaron en un estudio de 54 niños que se trataron con quimioterapia de inducción (por lo general, doxorrubicina, bleomicina, vincristina, etopósido, prednisona y quimioterapia de ciclofosfamida) y 21 Gy de radioterapia entre 2008 y 2016.[12]
Al cabo de una mediana de seguimiento de 39,5 meses, 3 de 54 pacientes (5,6 %) o 3 de 108 pulmones (2,8 %) presentaron neumonitis inducida por radiación (NR); 2 pulmones exhibieron NR de grado 1 y 1 pulmón exhibió NR de grado 2.
La NR se observó solo en los pacientes con una mediana de dosis pulmonar superior a 12,4 Gy o con una mediana de dosis pulmonar individual superior a 13,8 Gy.
En un estudio de 109 pacientes (59 % con linfoma de Hodgkin) que fueron irradiados a una mediana de edad de 13,8 años (intervalo, 0,04–20,9 años; mediana de seguimiento, 3,4 años), los pacientes se trataron con una dosis media de radioterapia prescrita de 21 Gy (intervalo 0,4–64,8 Gy); el 58,7 % de los pacientes recibió bleomicina y el 83,5 % de los pacientes recibió ciclofosfamida.[13]
La incidencia acumulada a 5 años después de la irradiación fue la siguiente: un 6 % de neumonitis; un 10 % de tos crónica; un 35 % de neumonía; un 11 % de disnea; un 2 % de requerimiento de oxígeno suplementario; un 40 % de enfermedad pulmonar intersticial y un 12 % de deformidad de la pared torácica.
Un paciente murió por insuficiencia respiratoria progresiva.
En las pruebas disponibles de funcionamiento pulmonar postirradiación de 44 pacientes se observaron indicios de enfermedad pulmonar obstructiva (25 %), enfermedad restrictiva (11 %), tórax distendido (32 %) y anomalías en la capacidad de difusión (12 %).
En el análisis univariante, la cirugía torácica, la bleomicina, la edad, la dosis máxima de irradiación pulmonar V30 (volumen de pulmón expuesto a una dosis de radiación >30 Gy), y el volumen de pulmón expuesto a radiación mayor a 25 Gy fueron factores de riesgo significativo para desenlaces pulmonares adversos.
Se demostró una relación entre la dosis media de irradiación pulmonar, la dosis máxima pulmonar y la dosis V (porcentaje de volumen pulmonar que recibe la dosis límite o superior). En análisis multivariante, la dosis media de irradiación pulmonar fue el único factor pronóstico significativo de un desenlace pulmonar adverso (P = 0,01).
Se notificaron cambios en el funcionamiento pulmonar en niños tratados con radioterapia dirigida a todo el pulmón para tratar un tumor de Wilms metastásico.[14,15]
Una dosis de 12 Gy a 14 Gy redujo la capacidad pulmonar total (CPT) y la capacidad vital del pulmón hasta casi el 70 % de los valores previstos, y aún menos en los pacientes sometidos a toracotomía.
La administración de bleomicina sola quizá produzca toxicidad pulmonar y, cuando se combina con radioterapia, a veces intensifica las reacciones a la radiación. Los fármacos quimioterapéuticos, como la doxorrubicina, la dactinomicina y el busulfano, son radiomiméticos y es posible que reactiven un daño latente causado por la radiación.[14,15]
Complicaciones respiratorias posteriores a la quimioterapia
Los fármacos quimioterapéuticos que causan toxicidad pulmonar y que se usan por lo general para el tratamiento de las neoplasias malignas infantiles son la bleomicina, el busulfano y las nitrosoureas (carmustina y lomustina). Estos fármacos inducen por sí mismos daños en los pulmones o potencian los efectos dañinos de la radiación dirigida al pulmón.
El tratamiento de modalidad combinada, que incluye quimioterapia pulmonar tóxica y radioterapia torácica, o cirugía en la pared del tórax o en el tórax, aumenta el riesgo de deterioro del funcionamiento pulmonar.[7]
Evidencia (desenlaces en cohortes tratadas con quimioterapia pulmonar tóxica):
La fibrosis pulmonar con enfermedad restrictiva permanente relacionada con la bleomicina depende de la dosis; por lo habitual ocurre con dosis mayores de 200 U/m2 a 400 U/m2, que es más alta que las dosis usadas en protocolos de tratamiento para las neoplasias malignas infantiles.[16,17]
Los regímenes pediátricos para tratar linfomas de Hodgkin con radioterapia y doxorrubicina, bleomicina, vinblastina y dacarbazina (ABVD) mostraron una incidencia significativa de disfunción pulmonar asintomática después del tratamiento; esta parece mejorar con el paso del tiempo.[18] Sin embargo, se notificó toxicidad pulmonar de grados 3 y 4 en el 9 % de los niños que recibieron 12 ciclos de ABVD seguidos de 21 Gy de radiación de campo ampliado.[17]
La toxicidad pulmonar relacionada con la administración de ABVD a veces produce fibrosis inducida por la bleomicina o la llamada neumonitis por evocación de la radiación, que se relaciona con la administración de doxorrubicina.
En una serie participaron 207 niños que recibieron tratamiento con bleomicina para linfoma de Hodgkin o tumores de células germinativas y se sometieron a pruebas de función pulmonar como mínimo 1 año después de finalizar el tratamiento. Al menos una anomalía de la función pulmonar estaba presente en el 52,5 % de los pacientes. Se observó enfermedad pulmonar obstructiva en el 22,7 % de los pacientes, enfermedad pulmonar restrictiva en el 7,5 %, hiperinsuflación en el 28,8 % y ventilación no uniforme en el 14 %. Muy pocos pacientes presentaron síntomas pulmonares.[19].[19]
La neumopatía venoclusiva se observó con poca frecuencia y se la atribuyó a la bleomicina o alquilantes.[20,21]
Complicaciones respiratorias relacionadas con un trasplante de células madre hematopoyéticas
Los pacientes sometidos a un TCMH tienen un mayor riesgo de toxicidad pulmonar, relacionada con los siguientes aspectos:[22]
Regímenes de acondicionamiento, incluso ciclofosfamida, busulfano o carmustina.
Irradiación corporal total (ICT).
Enfermedad de injerto contra huésped (EICH).
Pese a que la mayoría de los sobrevivientes no tienen compromiso clínico, en ocasiones se presenta una neumopatía restrictiva y se notificó mayor prevalencia con el aumento del tiempo a partir del TCMH, de acuerdo con los datos limitados de cohortes a las que se hizo un seguimiento longitudinal.[23,24]
La neumopatía obstructiva es menos común, así como el síndrome pulmonar de inicio tardío, que abarca la enfermedad restrictiva y la enfermedad obstructiva.
Se ha notificado enfermedad pulmonar restrictiva o intersticial en sobrevivientes de neuroblastoma de riesgo alto sometidos a un TCMH (con ITC o sin esta). En una serie de 48 sobrevivientes, la radioterapia no tuvo correlación directa con las pruebas de función pulmonar, mientras que la ciclofosfamida y la ifosfamida sí afectaron las pruebas de función pulmonar. Una dosis media más alta de ciclofosfamida se relacionó con enfermedad pulmonar restrictiva, mientras que dosis más altas de ifosfamida se correlacionaron tanto con enfermedad pulmonar restrictiva como con enfermedad pulmonar intersticial. En esta cohorte, el 25 % de los pacientes eran sintomáticos, pero no todos estos síntomas se correlacionaron con pruebas de función pulmonar anormales.[25]
A veces se presenta bronquiolitis obliterante, con neumonía organizada o sin ella, daño alveolar difuso y neumonía intersticial como uno de los componentes de este síndrome, por lo general entre 6 y 12 meses después del trasplante. Es posible que se presenten tos, disnea o sibilancias, con una radiografía normal o con imágenes de infiltrados difusos o en parches; no obstante, la mayoría de los pacientes son asintomáticos.[26,27]
Otros factores relacionados con los efectos tardíos respiratorios
Los factores adicionales que contribuyen a los efectos tóxicos pulmonares crónicos incluyen infecciones coincidentes, neumopatía subyacente (por ejemplo, asma), anomalías en la pared torácica, efectos tóxicos respiratorios, EICH crónica y los efectos del compromiso pulmonar crónico por un tumor o reacción a un tumor.[8]
La lobectomía pulmonar durante la niñez no parece tener un efecto importante en el funcionamiento pulmonar a largo plazo,[28] pero el efecto a largo plazo de la cirugía pulmonar en niños con cáncer no está bien definido.
Las complicaciones pulmonares quizás se exacerben por fumar cigarrillos u otras sustancias. Si bien las tasas de consumo de tabaco en los sobrevivientes de cáncer infantil tienden a ser más bajas que las de la población general, sigue siendo importante impedir el inicio del consumo de tabaco y promover su abandono en esta población definida.[29]
Evidencia (disfunción pulmonar en exfumadores o fumadores actuales):
Las evaluaciones del funcionamiento pulmonar de 433 adultos sobrevivientes de cáncer infantil tratados con modalidades tóxicas para los pulmones demostraron un riesgo significativamente más alto de disfunción pulmonar en los fumadores, en comparación con los no fumadores.[30]
Los valores medianos de FEV1 o CVF entre los fumadores actuales y los exfumadores fueron más bajos que los de las personas que nunca fumaron.
Los valores medianos de FEV1 o CVF fueron más bajos en quienes tenían un índice de consumo de tabaco de menos de 6 o de 6 paquetes o más por año, en comparación con quienes nunca habían fumado. Esto indica que los sobrevivientes que son exfumadores o fumadores activos tienen un aumento de riesgo de enfermedad pulmonar obstructiva y restrictiva en el futuro.
En el Cuadro 18 se resumen los efectos tardíos respiratorios y los exámenes médicos de detección correspondientes.
Cuadro 18. Efectos tardíos respiratoriosa
Tratamiento predisponente
Efectos respiratorios
Exámenes médicos de detección e intervenciones
CDCO = capacidad de difusión pulmonar del monóxido de carbono; EICH = enfermedad de injerto contra huésped; TCMH = trasplante de células madre hematopoyéticas.
Busulfano; carmustina (BCNU)/lomustina (CCNU); bleomicina; radioterapia con exposición de los pulmones y cirugía que afecta el funcionamiento pulmonar (lobectomía, metastasectomía, resección en cuña)
Antecedentes: tos, dificultad respiratoria, disnea de esfuerzo y sibilancias
Examen pulmonar
Pruebas del funcionamiento pulmonar (incluso CDCO y espirometría)
Radiografía de tórax
Orientación para evitar el tabaco o dejar de fumar
Para pacientes con pruebas de funcionamiento pulmonar o radiografía de tórax anormales, se debe sopesar si se repite la evaluación antes de administrar anestesia general
Consulta de neumología para pacientes con disfunción pulmonar sintomática
Antecedentes: tos, dificultad respiratoria, disnea de esfuerzo y sibilancias
Examen pulmonar
Pruebas del funcionamiento pulmonar (incluso CDCO y espirometría)
Radiografía de tórax
Orientación para evitar el tabaco o dejar de fumar
Para pacientes con pruebas de funcionamiento pulmonar o radiografía de tórax anormales, se debe sopesar si se repite la evaluación antes de administrar anestesia general
Consulta de neumología para pacientes con disfunción pulmonar sintomática
Smith L, Glaser AW, Peckham D, et al.: Respiratory morbidity in young people surviving cancer: Population-based study of hospital admissions, treatment-related risk factors and subsequent mortality. Int J Cancer 145 (1): 20-28, 2019. [PUBMED Abstract]
Fidler MM, Reulen RC, Bright CJ, et al.: Respiratory mortality of childhood, adolescent and young adult cancer survivors. Thorax 73 (10): 959-968, 2018. [PUBMED Abstract]
Armenian SH, Landier W, Francisco L, et al.: Long-term pulmonary function in survivors of childhood cancer. J Clin Oncol 33 (14): 1592-600, 2015. [PUBMED Abstract]
Dietz AC, Chen Y, Yasui Y, et al.: Risk and impact of pulmonary complications in survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Cancer 122 (23): 3687-3696, 2016. [PUBMED Abstract]
Green DM, Zhu L, Wang M, et al.: Pulmonary Function after Treatment for Childhood Cancer. A Report from the St. Jude Lifetime Cohort Study (SJLIFE). Ann Am Thorac Soc 13 (9): 1575-85, 2016. [PUBMED Abstract]
Huang TT, Hudson MM, Stokes DC, et al.: Pulmonary outcomes in survivors of childhood cancer: a systematic review. Chest 140 (4): 881-901, 2011. [PUBMED Abstract]
Mulder RL, Thönissen NM, van der Pal HJ, et al.: Pulmonary function impairment measured by pulmonary function tests in long-term survivors of childhood cancer. Thorax 66 (12): 1065-71, 2011. [PUBMED Abstract]
Kasteler R, Weiss A, Schindler M, et al.: Long-term pulmonary disease among Swiss childhood cancer survivors. Pediatr Blood Cancer 65 (1): , 2018. [PUBMED Abstract]
Josephson MB, Goldfarb SB: Pulmonary complications of childhood cancers. Expert Rev Respir Med 8 (5): 561-71, 2014. [PUBMED Abstract]
Briere TM, Agrusa JE, Martel MK, et al.: Acute and Late Pulmonary Effects After Radiation Therapy in Childhood Cancer Survivors: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 533-548, 2024. [PUBMED Abstract]
Venkatramani R, Kamath S, Wong K, et al.: Pulmonary outcomes in patients with Hodgkin lymphoma treated with involved field radiation. Pediatr Blood Cancer 61 (7): 1277-81, 2014. [PUBMED Abstract]
Lewis GD, Agrusa JE, Teh BS, et al.: Radiation pneumonitis in pediatric Hodgkin lymphoma patients receiving radiation therapy to the chest. Pract Radiat Oncol 8 (5): e364-e368, 2018 Sep - Oct. [PUBMED Abstract]
Venkatramani R, Kamath S, Wong K, et al.: Correlation of clinical and dosimetric factors with adverse pulmonary outcomes in children after lung irradiation. Int J Radiat Oncol Biol Phys 86 (5): 942-8, 2013. [PUBMED Abstract]
McDonald S, Rubin P, Maasilta P: Response of normal lung to irradiation. Tolerance doses/tolerance volumes in pulmonary radiation syndromes. Front Radiat Ther Oncol 23: 255-76; discussion 299-301, 1989. [PUBMED Abstract]
McDonald S, Rubin P, Phillips TL, et al.: Injury to the lung from cancer therapy: clinical syndromes, measurable endpoints, and potential scoring systems. Int J Radiat Oncol Biol Phys 31 (5): 1187-203, 1995. [PUBMED Abstract]
Bossi G, Cerveri I, Volpini E, et al.: Long-term pulmonary sequelae after treatment of childhood Hodgkin's disease. Ann Oncol 8 (Suppl 1): 19-24, 1997. [PUBMED Abstract]
Fryer CJ, Hutchinson RJ, Krailo M, et al.: Efficacy and toxicity of 12 courses of ABVD chemotherapy followed by low-dose regional radiation in advanced Hodgkin's disease in children: a report from the Children's Cancer Study Group. J Clin Oncol 8 (12): 1971-80, 1990. [PUBMED Abstract]
Hudson MM, Greenwald C, Thompson E, et al.: Efficacy and toxicity of multiagent chemotherapy and low-dose involved-field radiotherapy in children and adolescents with Hodgkin's disease. J Clin Oncol 11 (1): 100-8, 1993. [PUBMED Abstract]
De A, Guryev I, LaRiviere A, et al.: Pulmonary function abnormalities in childhood cancer survivors treated with bleomycin. Pediatr Blood Cancer 61 (9): 1679-84, 2014. [PUBMED Abstract]
Polliack A: Late therapy-induced cardiac and pulmonary complications in cured patients with Hodgkin's disease treated with conventional combination chemo-radiotherapy. Leuk Lymphoma 15 (Suppl 1): 7-10, 1995. [PUBMED Abstract]
Ranchoux B, Günther S, Quarck R, et al.: Chemotherapy-induced pulmonary hypertension: role of alkylating agents. Am J Pathol 185 (2): 356-71, 2015. [PUBMED Abstract]
Marras TK, Chan CK, Lipton JH, et al.: Long-term pulmonary function abnormalities and survival after allogeneic marrow transplantation. Bone Marrow Transplant 33 (5): 509-17, 2004. [PUBMED Abstract]
Inaba H, Yang J, Pan J, et al.: Pulmonary dysfunction in survivors of childhood hematologic malignancies after allogeneic hematopoietic stem cell transplantation. Cancer 116 (8): 2020-30, 2010. [PUBMED Abstract]
Frisk P, Arvidson J, Hedenström H: A longitudinal study of pulmonary function after stem cell transplantation, from childhood to young adulthood. Pediatr Blood Cancer 58 (5): 775-9, 2012. [PUBMED Abstract]
Hobbie WL, Li Y, Carlson C, et al.: Late effects in survivors of high-risk neuroblastoma following stem cell transplant with and without total body irradiation. Pediatr Blood Cancer 69 (3): e29537, 2022. [PUBMED Abstract]
Uderzo C, Pillon M, Corti P, et al.: Impact of cumulative anthracycline dose, preparative regimen and chronic graft-versus-host disease on pulmonary and cardiac function in children 5 years after allogeneic hematopoietic stem cell transplantation: a prospective evaluation on behalf of the EBMT Pediatric Diseases and Late Effects Working Parties. Bone Marrow Transplant 39 (11): 667-75, 2007. [PUBMED Abstract]
Yoshihara S, Yanik G, Cooke KR, et al.: Bronchiolitis obliterans syndrome (BOS), bronchiolitis obliterans organizing pneumonia (BOOP), and other late-onset noninfectious pulmonary complications following allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 13 (7): 749-59, 2007. [PUBMED Abstract]
Kreisel D, Krupnick AS, Huddleston CB: Outcomes and late complications after pulmonary resections in the pediatric population. Semin Thorac Cardiovasc Surg 16 (3): 215-9, 2004. [PUBMED Abstract]
Gibson TM, Liu W, Armstrong GT, et al.: Longitudinal smoking patterns in survivors of childhood cancer: An update from the Childhood Cancer Survivor Study. Cancer 121 (22): 4035-43, 2015. [PUBMED Abstract]
Oancea SC, Gurney JG, Ness KK, et al.: Cigarette smoking and pulmonary function in adult survivors of childhood cancer exposed to pulmonary-toxic therapy: results from the St. Jude lifetime cohort study. Cancer Epidemiol Biomarkers Prev 23 (9): 1938-43, 2014. [PUBMED Abstract]
Efectos tardíos en los órganos de los sentidos
Audición
La hipoacusia como efecto tardío del tratamiento se presenta después de recibir compuestos derivados del platino (cisplatino, carboplatino), radioterapia craneal o ambos. Estas exposiciones terapéuticas son más comunes durante el tratamiento de tumores sólidos en el sistema nervioso central (SNC) y fuera del SNC. Los niños son más susceptibles que los adultos a los efectos tóxicos otológicos de los derivados del platino.[1]
Evidencia (hipoacusia):
En un estudio longitudinal se evaluó la incidencia y los factores de riesgo de la hipoacusia en 171 pacientes (340 oídos) con tumores del SNC y de cabeza y cuello tratados con múltiples exposiciones ototóxicas. En el estudio se notificaron relaciones entre la pérdida de audición grave, la quimioterapia ototóxica y la dosis coclear.[2]
La media de la dosis coclear (oportunidad relativa [OR], 1,04 por Gy; P < 0,001), el tiempo desde la radioterapia (OR, 1,21 por año; P < 0,001), la dosis de cisplatino (OR, 1,48 por 100 mg/m2; P < 0,001) y la dosis de carboplatino (OR, 1,41 por 1.000 mg/m2;P = 0,002) se relacionaron con el aumento del grado de hipoacusia, según la escala de ototoxicidad de la International Society of Pediatric Oncology (SIOP)-Boston.
La incidencia acumulada de la pérdida de audición de alta frecuencia (>4 kHz) fue del 50 % o más 5 años después de la radiación cuando la media de la dosis coclear era superior a 30 Gy.
La incidencia de la pérdida de audición en todas las frecuencias siguió aumentando más allá de los 5 años después de la radioterapia.
No hubo ningún efecto sinérgico de la radioterapia y el cisplatino (término de interacción, P = 0,53) o la radioterapia y el carboplatino (término de interacción, P = 0,85).
En un informe del Swiss Childhood Cancer Survivor Study (CCSS) (n = 2061), se calculó que la prevalencia de hipoacusia en los sobrevivientes fue del 10 %, en comparación con el 3 % en los hermanos de ambos sexos. La hipoacusia fue más común en los sobrevivientes de tumores del SNC (25 %), neuroblastoma (23 %), tumor hepático (21 %), tumor de células germinativas (20 %), tumor óseo (16 %) y sarcoma de tejido blando (16 %).[3]
Un estudio de seguimiento de esta cohorte se limitó a 270 sobrevivientes de cáncer infantil tratados con quimioterapia a base de platino. Se utilizaron dosis variables de cisplatino. Entre los que recibieron cisplatino, la mediana de dosis acumulada fue de 400 mg/m2 (intervalo intercuartílico, 314–480 mg/m2). La pérdida de audición, determinada mediante evaluaciones audiológicas, se caracterizó como leve (20 %), moderada (29 %) y grave (28 %). La hipoacusia más grave se relacionó con una edad más temprana en el momento del diagnóstico (OR, 5,4 durante <5 años), tratamiento en años anteriores (OR, 4,8 para 1990–1995), dosis más altas de cisplatino (OR, 13,5 para >450 mg/m2), radioterapia craneal concomitante (OR, 4,4) y trasplante de células madre hematopoyéticas (TCMH) (OR, 2,7).[4]
En un estudio poblacional se usaron registros de datos sanitarios para evaluar la incidencia a largo plazo y los factores de predicción de hipoacusia que requiere audífonos entre sobrevivientes canadienses de cáncer infantil.[5]
Entre 11 842 casos y 59 210 controles identificados, los casos tuvieron mayor riesgo de necesitar audífonos (cociente de riesgos instantáneos [CRI], 12,8; Intervalo de confianza [IC] 95 %, 9,8–16,7).
La incidencia acumulada de uso de audífonos entre los sobrevivientes fue de 2,1 % (IC 95 %, 1,7–2,5 %) a los 20 años y aumentó a 6,4 % (IC 95 %, 2,8–12,1 %) 30 años después del diagnóstico.
La incidencia a 30 años más alta la presentaron los sobrevivientes de neuroblastoma (10.7 %; IC 95 %, 3,8–21,7%) y hepatoblastoma (16,2 %; IC 95 %, 8,6–26,0 %).
En análisis multivariantes los factores de predicción para la necesidad de audífonos fueron los siguientes:
Edad de 0 a 4 años en el momento del diagnóstico versus edad de 5 a 9 años (CRI, 2,2; IC 95 %, 1,4–3,3).
Tratamiento con 200 mg/m2 o más de cisplatino versus tratamiento sin cisplatino (CRI, 3,1; 1,8–5,5).
Tratamiento con radiación craneal o facial de más de 32 Gy versus radioterapia de 32 Gy o menor (CRI, 2,4; 1,6–3,7),
Los datos del North American Childhood Cancer Survivor Study (CCSS) indican que la tasa relativa de primera aparición de complicaciones auditivas (es decir, problemas de audición, acúfenos, hipoacusia, sordera) es más alta en el período comprendido entre el diagnóstico y los 5 años posteriores; sin embargo, durante los 5 años que siguen al diagnóstico, el riesgo de estas afecciones en los sobrevivientes permaneció significativamente más alto que para los hermanos de ambos sexos.[6]
Los factores de riesgo relacionados con la hipoacusia son los siguientes:
Edad más temprana en el momento del tratamiento.[5,7]
Dosis acumuladas más altas de quimioterapia a base de platino (≥200 mg/m2).[5,8] De acuerdo con los resultados de un estudio de cohorte multiinstitucional en América del Norte (n = 1481), los factores de riesgo significativos para presentar hipoacusia inducida por cisplatino fueron la dosis acumulada de cisplatino y la forma de administración del medicamento. La administración de dosis fraccionadas más altas se vinculó con un riesgo alto de hipoacusia inducida por cisplatino (por cada 10 mg/m2 de aumento/día: OR ajustada, 1,15; por cada 50 mg/m2 aumento por ciclo: OR ajustada, 2,16).[7]
Exposición al cisplatino combinado con carboplatino.[8]
Hipoacusia y tratamiento con derivados del platino
La hipoacusia neurosensorial relacionada con derivados del platino se presenta como toxicidad aguda que, por lo general, es irreversible y bilateral.[9]
La prevalencia de hipoacusia ha variado mucho por serie y depende del tratamiento con derivados del platino (por ejemplo, el tipo de derivado del platino, la dosis y la duración de la infusión), los factores del huésped (por ejemplo, edad, susceptibilidad genética y funcionamiento renal), la administración de tratamiento ototóxico adicional (radioterapia craneal, aminoglucósidos y diuréticos de asa) y los criterios de clasificación utilizados para notificar la prevalencia y gravedad de la pérdida de audición.[8-10]
La hipoacusia se manifiesta al inicio en las frecuencias altas y progresa a las frecuencias del habla a medida que aumenta la exposición acumulada.[9]
En pacientes pediátricos, la hipoacusia inducida por cisplatino que compromete las frecuencias del habla (500–2000 Hz) se suele presentar con dosis acumuladas que exceden 400 mg/m2.[11,12]
Se notificó que la prolongación de la infusión o el fraccionamiento de la dosis reducen el riesgo de hipoacusia importante.[13]
En 2 ensayos aleatorizados (Children's Oncology Group ACCL0431 [NCT00716976] e International Childhood Liver Tumour Strategy Group SIOPEL-6 [NCT00652132]), se demostró la eficacia del tiosulfato de sodio para prevenir la pérdida de audición inducida por el cisplatino.[14,15]
En el ensayo SIOPEL-6 se comparó el cisplatino solo con el cisplatino con la administración retardada de tiosulfato sódico (tiosulfato sódico administrado 6 horas después del cisplatino) y se observó una incidencia un 48 % menor de pérdida de audición inducida por cisplatino entre los niños con hepatoblastoma de riesgo estándar. El tratamiento con tiosulfato sódico no puso en peligro la supervivencia general ni la supervivencia sin complicaciones.[15] Estos ensayos llevaron a la aprobación reglamentaria del tiosulfato de sodio para la prevención de la pérdida de audición inducida por cisplatino. Sin embargo, cada ensayo usó un criterio de valoración de la audición diferente. En 2010, se creó la escala de ototoxicidad SIOP-Boston para superar las limitaciones más importantes de las escalas disponibles; se considera que tiene un papel fundamental en la armonización de los criterios de valoración de la audición. Mediante la escala SIOP, se reevaluaron los resultados auditivos del ensayo ACCL0431. Este análisis demostró que los niños tratados con tiosulfato sódico tenían alrededor de un 90 % menos de probabilidades de presentar una pérdida de audición inducida por cisplatino superior al grado 2 durante el período del ensayo o al final del tratamiento con cisplatino, en comparación con el grupo de observación.[16]
Se notificó que los efectos ototóxicos posteriores a la quimioterapia con derivados del platino empeoran a medida que transcurren los años después de terminar el tratamiento.[17]
La radioterapia dirigida a la fosa posterior, que incluye el 8.o nervio craneal (que indica daño cloquear al final del tratamiento), aumenta el riesgo de hipoacusia de inicio tardío en los sobrevivientes tratados con cisplatino.[18]
Por lo general, el uso de carboplatino en dosis convencionales (no mielodepresoras) no es ototóxico.[19] No obstante, se notificó hipoacusia de aparición tardía en las siguientes poblaciones:
En un análisis transversal y multicéntrico que incluyó a 451 sobrevivientes neerlandeses de cáncer infantil que recibieron fármacos derivados del platino pero no radioterapia craneal, la incidencia de ototoxicidad (definida como grado de Münster >2b [>20dB a ≥4–8kHz]) vinculada con el uso de carboplatino administrado solo (n = 112) fue del 17 %.[8]
En un estudio único de efectos tóxicos otológicos después de la dosificación de carboplatino para trasplantes diferentes al de TCMH en pacientes con retinoblastoma, se notificó que 8 de 175 niños presentaron hipoacusia. En 7 de los 8 niños, la aparición de los efectos tóxicos otológicos se retrasó por una mediana de 3,7 años.[20]
En otro estudio en el que se evaluaron los desenlaces audiológicos en 60 sobrevivientes de retinoblastoma tratados con carboplatino sistémico no mielosupresor y vincristina, se calculó una incidencia acumulada de hipoacusia del 20,3 % a los 10 años.[21]
Entre los 10 pacientes (17 %) que presentaron hipoacusia persistente de grado 3 o grado 4, en el momento de iniciar la quimioterapia 9 eran menores de 6 meses de edad.
Una edad menor al inicio del tratamiento fue el único factor pronóstico significativo de hipoacusia; la incidencia acumulada de hipoacusia fue del 39 % en los pacientes menores de 6 meses versus solo el 8,3 % en los pacientes de 6 meses o más.
Es posible que el uso de carboplatino como régimen de acondicionamiento para un TCMH, en particular combinado con tratamiento previo con carboplatino o cisplatino, cause efectos otológicos importantes.[11,12]
En una serie se evaluó a 115 pacientes con tumores de células germinativas recidivantes o resistentes al tratamiento (mediana de edad, 32 años) que habían recibido quimioterapia de primera línea a base de cisplatino. En el estudio se informó de los resultados auditivos de adolescentes y adultos jóvenes tratados con 3 ciclos de terapia de carboplatino a dosis altas. De los 106 pacientes con audición normal o pérdida de audición leve antes de la terapia con carboplatino a dosis altas, 70 (66 %) presentaron una pérdida de audición de moderada a grave en las frecuencias del habla tras la terapia con carboplatino a dosis altas. A 25 pacientes (22 %) se les recomendó el uso de audífonos tras el tratamiento con carboplatino a dosis altas. Los pacientes con pérdida de audición de moderada a grave aislada en frecuencias altas antes del tratamiento con carboplatino a dosis altas tenían más probabilidades de presentar una pérdida de audición de moderada a grave en las frecuencias del habla después del tratamiento y de que se les recomendaran el uso de audífonos.[22]
Hipoacusia y radioterapia craneal
Cuando se usa como modalidad simple, es posible que la radioterapia craneal produzca efectos ototóxicos que a veces se presentan en forma gradual y se manifiestan meses o años después de la exposición. La dosis liminar que produce toxicidad auditiva después de la radioterapia sola se ubica en el intervalo de 35 a 45 Gy para los niños.[23]
La hipoacusia neurosensorial de las frecuencias altas no es habitual si las dosis acumuladas de radiación son inferiores a 35 Gy y, casi nunca es grave con dosis inferiores a 45 Gy.[24] La excepción se presenta en pacientes con tumores supratentoriales y derivaciones ventriculoperitoneales, para quienes dosis inferiores a 30 Gy a veces se relacionan con hipoacusia en las frecuencias intermedias (1000–2000 Hz).[23,25]
Para reducir el riesgo de hipoacusia, la dosis coclear promedio no debe exceder 30 Gy a 35 Gy, administrados durante más de 6 semanas.
La hipoacusia neurosensorial después de radioterapia craneal quizá empeore con el tiempo.
En un estudio de 235 pacientes con tumores de encéfalo infantiles tratados con radioterapia conformada o de intensidad modulada (sin cisplatino ni hipoacusia previa) y sometidos a vigilancia durante una mediana de 9 años, la hipoacusia neurosensorial fue prevalente en el 14 % de los pacientes, con una mediana de tiempo hasta el inicio de 3,6 años a partir de la radioterapia.[26]
En las evaluaciones de seguimiento de 29 pacientes identificados se observó un empeoramiento continuado de la sensibilidad auditiva.
Los factores de riesgo de hipoacusia neurosensorial relacionada con radiación craneal fueron menor edad al inicio de la radiación, mayor dosis de radiación coclear y derivación del líquido cefalorraquídeo.
La radioterapia administrada en forma simultánea con el cisplatino puede exacerbar en forma considerable la hipoacusia relacionada con la quimioterapia con derivados del platino.[23,27-29]
En dos informes del CCSS, se notificaron los siguientes factores clínicos y genéticos en sobrevivientes de cáncer a 5 años:
Mayor riesgo de problemas auditivos (riesgo relativo [RR], 2,3), acúfenos (RR, 1,7), hipoacusia para la que se necesitó un audífono (RR, 4,4) e hipoacusia en uno o ambos oídos que no se corrigió con un audífono (RR, 5,2), en comparación con los hermanos de ambos sexos. La radioterapia dirigida al lóbulo temporal (>30 Gy) y la fosa posterior (>50 Gy, pero también 30–49,9 Gy) guardó relación con estos desenlaces adversos.[6]
La exposición a derivados del platino se relacionó con un aumento de riesgo de problemas de audición (RR, 2,1), acúfenos (RR, 2,8) e hipoacusia para la que se necesitó un audífono (RR, 4,1).[6]
Los hombres tuvieron más propensión que las mujeres a notificar acúfenos relacionados con la radioterapia craneal (9,4 vs. 5,4 %) y pérdida de audición (14,0 vs. 10,7 %).[30]
Los sobrevivientes con acúfenos o pérdida de audición tuvieron más propensión que los controles a presentar mareos o vértigos persistentes, tomar antidepresivos y notificar un estado de salud general más precario.[30]
En un estudio de asociación del genoma completo se identificó un polimorfismo de un solo nucleótido en el cromosoma 6 como casi significativo en todo el genoma para la asociación con la pérdida de audición relacionada con la radioterapia craneal. En un análisis de replicación del estudio de cohorte St. Jude Life (SJLIFE) se confirmó la relación significativa con la pérdida de audición relacionada con la radioterapia craneal y de novo.[30]
La investigación del Pediatric Normal Tissue Effects in the Clinic (PENTEC) incluyó 457 cócleas en riesgo de pacientes tratados con radioterapia sin quimioterapia, y 398 cócleas de pacientes tratados con quimioterapia.[31]
La incidencia y la gravedad de la pérdida de audición coclear por exposición exclusiva a radioterapia están relacionadas con la dosis y la edad. El riesgo de pérdida de audición fue inferior al 5 % en la cóclea que recibió una dosis media de 35 Gy o inferior, pero aumentó hasta el 30 % con una dosis de 50 Gy.
El riesgo de pérdida de audición osciló entre el 25 % y el 40 % en niños menores de 5 años en el momento del diagnóstico, disminuyendo hasta el 10 % en niños mayores para cualquier dosis de radiación.
La probabilidad de sufrir una pérdida de audición grave similar a dosis de 18,3 Gy fue superior en los niños menores de 3 años que en los mayores de esa edad.
La hipoacusia de alta frecuencia fue la más frecuente, con una media de aparición de 3,6 años (intervalo, 0,4–13,2 años) después de la radioterapia.
También se observó pérdida de audición de baja frecuencia tras la radioterapia (en contraste con la quimioterapia, que afecta de forma principal a la alta frecuencia).
Se evaluaron las comorbilidades crónicas en una cohorte de 6778 sobrevivientes de 2 años de cáncer en adolescentes y adultos jóvenes (AAJ) (definidos como personas diagnosticadas de cáncer entre los 15 y los 39 años de edad), con una mediana de seguimiento de 5,1 años tras el diagnóstico de cáncer. El riesgo de presentar comorbilidades crónicas se comparó con el de personas emparejadas sin antecedentes de cáncer. La tasa de incidencia (TIR) bruta fue de 3,0 para la pérdida de audición relacionada con una exposición a la radiación superior a 30 Gy en la cabeza. En los análisis multivariantes, una exposición combinada a platino (>450 mg/m2) y radiación a la cabeza (30 Gy) se asoció con un mayor riesgo de pérdida de audición (TIR, 14,9).[32]
Hipoacusia y calidad de vida
Los niños tratados por neoplasias malignas tienen riesgo de hipoacusia de inicio temprano o tardío que, en ocasiones, afecta el aprendizaje, la comunicación, el rendimiento escolar, la interacción social y la calidad de vida en general.
La sensibilidad auditiva se mide mediante audiometría de tonos puros. Esta evaluación no mide el efecto social y emocional de la pérdida de audición. El efecto funcional de la pérdida de audición inducida por el tratamiento en la calidad de vida se examinó en adultos sobrevivientes de cáncer infantil del estudio de cohortes SJLIFE. Estos sobrevivientes recibieron quimioterapia a base de platino o radioterapia dirigida a la cabeza o el cuello y presentaban pérdida de audición documentada (n = 237).[33] Se evaluó a los participantes mediante la escala del Hearing Handicap Inventory for Adults. Los sobrevivientes con pérdida de audición grave (n = 156) tenían más probabilidades de sufrir aislamiento social (OR, 8,76), depresión (OR, 9,11) y ansiedad (OR, 17,57), además de tener ingresos personales reducidos (OR, 2,82) y menos empleos a tiempo completo (OR, 2,47). Los sobrevivientes que no utilizaban un audífono recomendado tenían el doble de probabilidades de tener un empleo que no era de tiempo completo (OR, 2,26) y unos ingresos personales reducidos (OR, 2,24) que los sobrevivientes que utilizaban audífono.
Entre 137 niños sobrevivientes de neuroblastoma (de 8 a 17 años), la hipoacusia se relacionó con problemas de lectura y las aptitudes para matemáticas, así como un riesgo más alto de dificultad de aprendizaje o necesidades de educación especial. Además, la hipoacusia se vinculó con una calidad de vida más precaria en relación con la escuela.[34]
Se realizaron evaluaciones neurocognitivas y de audiología seriadas en 260 niños y adultos jóvenes con tumores de encéfalo embrionarios inscritos en un protocolo de tratamiento que incluyó cirugía, irradiación craneoespinal adaptada al riesgo y quimioterapia. Los 64 niños con hipoacusia neurosensorial grave tuvieron mayores dificultades para la lectura con el tiempo en comparación con el grupo de niños con hipoacusia neurosensorial normal o leve a moderada. En particular, estos niños con hipoacusia neurosensorial grave parecían tener más dificultades con la capacidad fonológica y la velocidad de procesamiento, las cuales inciden en competencias de nivel superior, como la comprensión lectora.[35]
En un estudio de adultos sobrevivientes de tumores del SNC infantiles (n = 180) y de tumores sólidos fuera del SNC (n = 226) que recibieron tratamiento del cáncer potencialmente ototóxico, la hipoacusia grave (que exigió audífono o produjo sordera) se relacionó con un riesgo doble de dependencia, desempleo o de no terminar la escuela secundaria.[36]
El Children's Oncology Group publicó recomendaciones para la evaluación y el tratamiento de la hipoacusia en sobrevivientes de cánceres infantiles y cánceres en la adolescencia con el fin de promover la identificación temprana de los sobrevivientes en riesgo y la derivación oportuna a servicios correctivos.
En el Cuadro 19 se resumen los efectos tardíos auditivos y los exámenes médicos de detección correspondientes.
Amplificación en pacientes con hipoacusia progresiva
Terapia del habla y el lenguaje para los niños con hipoacusia
Consulta otorrinolaringológica para pacientes con infección crónica, tapón de cerumen u otros problemas anatómicos que exacerben o contribuyan a la hipoacusia
Adaptación de la infraestructura educativa (por ejemplo, asientos preferenciales en el aula, sistema de amplificación de FM, etc.)
Efectos orbitarios y ópticos
Las complicaciones orbitarias son comunes después de la radioterapia para retinoblastoma, sarcomas de cabeza y cuello, tumores del SNC e irradiación corporal total (ICT).
Retinoblastoma
Se ha avanzado en el tratamiento del retinoblastoma con mejores implantes para la enucleación, quimiorreducción intravenosa y quimioterapia intraarterial, además de quimioterapia intravítrea, termoterapia, crioterapia y radioterapia con placas.[37,38] Se requiere un seguimiento más prolongado para evaluar el efecto en la visión de los pacientes sometidos a estas modalidades de tratamiento más actuales.
Con anterioridad, los tumores ubicados cerca de la mácula y la fóvea se relacionaban con un mayor riesgo de complicaciones que conducían a la pérdida de la visión, aunque se observó que el tratamiento de estos tumores con ablación de la fóvea mediante láser es prometedor para conservar la visión.[39,40]
En un estudio de 61 pacientes (94 ojos) con una mediana de seguimiento de 51,8 meses, una dosis media de 7 Gy en el cristalino se relacionó con una incidencia a 5 años de cataratas del 20 % al 25 %.[41]
En un estudio de 64 ojos conservados de 42 pacientes (22 casos bilaterales) con retinoblastoma, al cabo de una media de seguimiento de 8 años, se identificó el estrabismo como secuela adversa muy frecuente y el 69 % de los pacientes tenían algún tipo de desviación. El estrabismo divergente (43 %) fue más común. El único factor de riesgo significativo fue la presencia de un tumor en la fóvea en el momento de la presentación inicial.[42]
En los sobrevivientes de retinoblastoma, la enucleación o la radioterapia pueden producir un volumen orbitario pequeño.
La edad inferior a 1 año quizás aumente el riesgo de complicaciones orbitarias, pero este hallazgo no es consistente en todos los estudios.[43,44]
En un análisis cualitativo de respuestas de encuestas de 404 sobrevivientes de retinoblastoma (mediana de tiempo desde el diagnóstico, 42 años) sometidos a enucleación, se encontraron efectos psicosociales relacionados con dificultades físicas e interpersonales derivadas de la apariencia, además de problemas sociales y de relaciones, en especial, burlas y acoso.[45]
El grupo de trabajo ocular del PENTEC revisó los factores dosimétricos y clínicos para cuantificar la dependencia de la dosis de radiación de los efectos adversos oculares tardíos, como retinopatía, neuropatía óptica y cataratas, en sobrevivientes de cáncer infantil que recibieron radioterapia craneal.[46]
Con base en datos limitados, el modelo para la presentación de retinopatía indica un riesgo de toxicidad del 5 % y del 50 % a 42 Gy y 62 Gy, respectivamente.
El modelo para la presentación de neuropatía óptica indica un riesgo de toxicidad del 5 % y del 50 % a 57 Gy y 64 Gy, respectivamente.
Los modelos para cataratas indican un riesgo del 5 % y del 50 % de cataratas autodeclaradas a 12 Gy y a más de 40 Gy, respectivamente, y un riesgo del 50 % de cataratas diagnosticadas por oftalmólogos a 9 Gy (>5 % de riesgo a largo plazo a 0 Gy en pacientes tratados con quimioterapia sola).
Los sobrevivientes de rabdomiosarcoma orbitario tienen riesgo de xeroftalmía, cataratas, hipoplasia orbitaria, ptosis, retinopatía, queratoconjuntivitis, neuropatía óptica, epitelioma del párpado y deterioro de la visión después de dosis de radioterapia de 30 Gy a 65 Gy.[47,48] A pesar de la precisión dosimétrica de la terapia de protones, se ha notificado la aparición de asimetría orbitaria tras una eficacia biológica relativa (EBR) superior a 40 Gy en múltiples huesos del reborde orbitario.[49]
En las últimas 3 décadas, las técnicas de tratamiento con radioterapia han evolucionado. Estas técnicas se examinaron en un estudio multinacional y transatlántico de sobrevivientes de rabdomiosarcoma de cabeza y cuello en los que con probabilidad se produjo deformidad facial como resultado del control local. En función de la localización del tumor, se observó una reducción del crecimiento facial en los sobrevivientes con tumores primarios no parameníngeos (P = 0,002) y tumores primarios parameníngeos (P < 0,001), pero no en los tumores primarios orbitarios (P = 0,080), en comparación con los controles sanos. En las 4 estrategias diferentes de terapia local utilizadas (terapia de protones, terapia de fotones, cirugía con radioterapia y cirugía con braquiterapia), el control local y la supervivencia fueron comparables. Al comparar la radioterapia con la cirugía y la braquiterapia, los pacientes orbitales presentaron más deformaciones faciales.[50]
Los intervalos de dosis de radioterapia más alta (>50 Gy) se relacionan con epiteliomas del párpado, queratoconjuntivitis, atrofia del conducto lagrimal y xeroftalmía grave.
Es posible que se presente retinitis y neuropatía óptica con dosis de 50 Gy a 65 Gy y hasta con dosis totales más bajas si el tamaño de la fracción individual es superior a 2 Gy.[51]
Se notificaron cataratas después de dosis más bajas, de 10 Gy a 18 Gy.[47]
Los sobrevivientes de glioma de la vía óptica también tienen riesgo de complicaciones visuales que obedecen, en parte, a la proximidad del tumor al nervio óptico.
En un estudio de cohortes retrospectivas con 59 pacientes pediátricos que presentaban gliomas de la vía óptica esporádicos diagnosticados entre 1990 y 2014 (mediana de seguimiento, 5,2 años), la carga de deterioro visual a largo plazo fue considerable.[52]
Más de dos tercios de los pacientes exhibían indicios de pérdida de visión a largo plazo, más de la mitad presentaba pérdida grave de visión en por lo menos un ojo y un cuarto de los pacientes tenía pérdida grave de visión bilateral.
Los factores de riesgo identificados para el desenlace visual precario fueron compromiso posquiasmático, edad temprana y palidez del nervio óptico en el momento de la presentación inicial.
El seguimiento longitudinal (mediana de 9 años) de 21 pacientes con gliomas de la vía óptica indicó que, antes del tratamiento, el 81 % de los pacientes presentaba reducción de la agudeza visual, el 81 % exhibía palidez del nervio óptico y todos presentaban reducción de los potenciales de evocación visual en uno o ambos ojos.[53]
El tratamiento detuvo la pérdida de agudeza visual durante 4 a 5 años.
La agudeza visual se mantuvo estable o mejoró en el 33 % de los pacientes en el momento del último seguimiento; sin embargo, en promedio disminuyó. La agudeza visual en el momento del seguimiento se relacionó con el volumen tumoral en la presentación inicial.
En un estudio de 51 niños con gliomas de grado bajo y tumores glioneurales de grado bajo diagnosticados durante el primer año de vida, la agudeza visual disminuyó en 27 de 48 pacientes (56 %), 13 (27 %) de los cuales se consideraron legalmente ciegos. La localización del tumor (hipotalámica o de la vía óptica) se relacionó de manera significativa con la disminución de la agudeza visual (P = 0,002).[54]
Los investigadores del CCSS evaluaron el efecto de las alteraciones visuales en los desenlaces cognitivos y psicosociales de 1233 adultos sobrevivientes de glioma infantil de grado bajo.[55]
En el 22,5 % de los pacientes se encontró algún grado de alteración visual; asimismo, el 3,8 % de los pacientes sufrían de ceguera bilateral.
Fue más probable que los sobrevivientes con ceguera bilateral estuvieran solteros, vivieran de forma dependiente y estuvieran desempleados que aquellos sin deterioro de la visión.
La ceguera bilateral no afectó los desenlaces cognitivos o emocionales. El deterioro de la visión (con algo de visión residual) no se relacionó con desenlaces psicológicos o económicos.
Efectos específicos del tratamiento
Los sobrevivientes de cáncer infantil tienen un aumento de riesgo de efectos tardíos oculares relacionados con la exposición del ojo tanto a glucocorticoides como a la radiación.
Evidencia (desarrollo de cataratas a partir de la exposición a la radiación):
En el CCSS se informó del aumento del riesgo de presentar efectos en la visión tardíos en los sobrevivientes a 5 años o más del diagnóstico. Estos riesgos tardíos incluían un aumento de riesgo de cataratas (RR, 10,8), glaucoma (RR, 2,5), ceguera legal (RR, 2,6), visión doble (RR, 4,1) y xeroftalmía (RR, 1,9), en comparación con sus hermanos.[56] En otro informe, los autores aportaron datos adicionales sobre el intervalo desde la radioterapia y la dosis de radiación relacionada con la presentación de cataratas.[57] Entre los 13 902 participantes en el estudio, el 3,5 % presentó cataratas (el 41 % en los 5 años siguientes a la radioterapia), con una mediana de tiempo hasta la aparición de 9,6 años y un tiempo máximo de 37 años. Las dosis de radiación sobre el cristalino se asociaron a un aumento de la prevalencia: 1,3% si era inferior a 0,5 Gy, 6,1 % después de 2,5 a 3,49 Gy, y 40,6 % después de 20 a 60 Gy.
Las dosis más altas se relacionaron con un intervalo de tiempo más corto hasta el diagnóstico. Del grupo de cataratas, el 31 % notificó haberse sometido a cirugía de cataratas, lo que corrobora las consecuencias clínicas.
El arabinósido de citosina (OR, 1,5) y la doxorrubicina (OR, 1,5) se asociaron de forma independiente con la presentación de cataratas, el metotrexato se relacionó de forma inversa (OR, 0,6) y no se observó ninguna interacción positiva entre el uso de corticosteroides y la radioterapia.
En una cohorte de 517 sobrevivientes de leucemia linfoblástica aguda infantil, la incidencia acumulada de cataratas a los 15 años fue del 4,5 % (mediana de 10,9 años desde el diagnóstico), según la evaluación sistemática mediante exámenes con lámpara de hendidura. La radioterapia dirigida al SNC fue el único factor de riesgo de cataratas relacionado con el tratamiento, que se presentó en el 11,1 % de los sobrevivientes sometidos a radiación, en comparación con el 2,8 % de quienes no la recibieron.[58]
La disminución de la agudeza visual es poco frecuente después de la radioterapia para las neoplasias intracraneales en los niños.
Evidencia (disminución de la agudeza visual debido a la exposición a la radiación):
En un estudio retrospectivo de 458 niños (875 ojos en riesgo) tratados con terapia de protones para una neoplasia intracraneal se evaluó el deterioro visual relacionado con la radioterapia. Se consideró que los ojos estaban en riesgo si el nervio óptico ipsilateral o el quiasma óptico recibieron 30 GyRBE o más de 0,1 cm3.[59]
Ningún paciente experimentó pérdida de visión completa.
La tasa actuarial a 5 años de disminución de la agudeza visual fue del 2,6 %.
La ubicación del tumor se correlacionó de manera significativa con el riesgo. Los investigadores observaron que los niños con tumores de la región supraselar o de la vía óptica corren mayor riesgo. El riesgo notificado fue de alrededor de un 5 % de disminución de la agudeza visual con una dosis de 56 GyRBE en el nervio óptico o la vía óptica.
En el caso de los niños con tumores en otras partes del encéfalo, no se observó ninguna toxicidad.
Se evaluaron las comorbilidades crónicas en una cohorte de 6778 sobrevivientes de 2 años de cáncer en adolescentes y adultos jóvenes (AAJ) (definidos como personas diagnosticadas de cáncer entre los 15 y los 39 años de edad), con una mediana de seguimiento de 5,1 años tras el diagnóstico de cáncer. El riesgo de presentar comorbilidades crónicas se comparó con el de personas emparejadas sin antecedentes de cáncer. La TIR bruta fue de 8,1 para la pérdida de visión asociada a una exposición a la radiación dirigida a la cabeza superior a 30 Gy.[32]
Las complicaciones oculares, como cataratas y xeroftalmía son comunes después del TCMH en la niñez.
Evidencia (efectos oculares del TCMH):
En una investigación del St. Jude Children's Research Hospital se evaluaron las complicaciones oculares de niños en edad escolar y adolescentes sobrevivientes durante por lo menos 1 año después de un TCMH alogénico. Entre 162 pacientes (media de edad en el momento del trasplante, 13,4 años; media de seguimiento, 4 años), se notificaron los siguientes resultados:[60]
Se observó formación de cataratas en 57 pacientes (35 %) y xeroftalmía 51 pacientes (31 %).
En el análisis multivariante de variables significativas se observó que la ICT fue un factor de riesgo de formación de cataratas, y la enfermedad de injerto contra huésped (EICH) crónica fue un factor de riesgo significativo para la xeroftalmía.
En comparación con los pacientes tratados con busulfano u otra quimioterapia, los pacientes tratados con una sola dosis o dosis fraccionadas de ICT tienen un aumento de riesgo de presentar cataratas.[61-64]
El riesgo oscila, en forma aproximada, entre el 10 % y el 60 % a los 10 años del tratamiento, dependiendo de la dosis total y el fraccionamiento, con un período de latencia más corto y cataratas más graves que se observan después de una fracción única y una tasa de dosis más alta de ICT.
En una serie se evaluó a 48 pacientes con neuroblastoma de riesgo alto (mediana de tiempo desde el diagnóstico, 14,1 años) que recibieron tratamiento con terapia multimodal, incluso quimioterapia a dosis altas, TCMH (con ICT o sin esta), radioterapia e inmunoterapia.[10]
De los sobrevivientes, 25 (52%) presentaron cataratas.
Todos los pacientes con cataratas habían recibido ICT (P < 0,0001).
Los pacientes que reciben dosis de ICT inferiores a 40 Gy tienen una probabilidad menor del 10 % de presentar cataratas graves.[64]
Los corticoesteroides y la enfermedad de injerto contra huésped (EICH) aumentan aún más el riesgo.[61,65]
En el programa Leucémie de l'Enfant et de l'Adolescent (LEA), la prevalencia de cataratas evaluada por pruebas seriadas con lámpara de hendidura en 271 participantes (mediana de seguimiento de 10,3 años) fue del 41,7 %; el 8,1 % necesitó intervención quirúrgica.[66]
La incidencia acumulada de cataratas entre aquellos tratados con ICT aumentó con el tiempo desde el 30 % a los 5 años hasta el 70,8 % a los 15 años y el 78 % a los 20 años.
La falta de estabilización en la incidencia de cataratas indica que casi todos los pacientes tratados con ICT presentarán cataratas a medida que aumente el período de seguimiento.
En contraste, la incidencia acumulada de cataratas a los 15 años fue del 12,5 % entre aquellos acondicionados con busulfano.
En un análisis multivariante, se identificó que una dosis acumulada alta de corticoesteroides era un posible cofactor de la ICT para el riesgo de cataratas.
Se ha observado que la xeroftalmia es la manifestación más común de EICH ocular, pero es posible que suceda como fenómeno aislado ante la ausencia de EICH
En un estudio de un solo centro se evaluó a 484 niños de manera consecutiva que recibieron un TCMH y a los que se realizaron exámenes oftalmológicos antes del TCMH y luego cada año hasta los 5 años posteriores al TCMH.[67]
La incidencia acumulada de EICH ocular crónica fue del 6,0 %.
La incidencia acumulada de enfermedad ocular seca de nueva aparición fue del 8,7 %.
La menor edad (0–4 años frente a 10–16 años) se relacionó con un menor riesgo de xeroftalmía.
En el Cuadro 20 se resumen los efectos tardíos oculares y los exámenes médicos de detección correspondientes.
Cuadro 20. Efectos tardíos ocularesa
Tratamiento predisponente
Efectos oculares o visuales
Exámenes médicos de detección e intervenciones
EICH = enfermedad de injerto contra huésped; TCMH = trasplante de células madre hematopoyéticas; 131I = yodo I 131.
Busulfano; corticoesteroides; radioterapia con exposición del ojo
Cataratas
Antecedentes: disminución de la agudeza visual, halos, diplopía
Hallazgos del examen ocular: agudeza visual y oftalmoscopia (anual)
Consulta oftalmológica
Radioterapia con exposición del ojo, incluso yodo radiactivo (131I)
Toxicidad ocular (hipoplasia orbitaria, atrofia del conducto lagrimal, xeroftalmia [queratoconjuntivitis seca], queratitis, telangiectasias, retinopatía, neuropatía del quiasma óptico, enoftalmia, dolor crónico en el ojo, maculopatía, papilopatía, glaucoma)
Antecedentes: cambios en la visión (disminución de la agudeza visual, halos, diplopía), xeroftalmía, irritación constante del ojo, lagrimeo excesivo, fotofobia, mala visión nocturna y dolor en el ojo
Hallazgos del examen ocular: agudeza visual y oftalmoscopia (anual)
Consulta oftalmológica
TCMH con cualquier antecedente de EICH crónica
Xeroftalmía (queratoconjuntivitis seca)
Antecedentes: xeroftalmía (ardor, picazón, sensación de cuerpo extraño e inflamación)
Hallazgos del examen ocular: agudeza visual y oftalmoscopia (anual)
Enucleación
Deterioro cosmético, ajuste precario de la prótesis, hipoplasia orbitaria
Li Y, Womer RB, Silber JH: Predicting cisplatin ototoxicity in children: the influence of age and the cumulative dose. Eur J Cancer 40 (16): 2445-51, 2004. [PUBMED Abstract]
Keilty D, Khandwala M, Liu ZA, et al.: Hearing Loss After Radiation and Chemotherapy for CNS and Head-and-Neck Tumors in Children. J Clin Oncol 39 (34): 3813-3821, 2021. [PUBMED Abstract]
Weiss A, Sommer G, Kasteler R, et al.: Long-term auditory complications after childhood cancer: A report from the Swiss Childhood Cancer Survivor Study. Pediatr Blood Cancer 64 (2): 364-373, 2017. [PUBMED Abstract]
Strebel S, Mader L, Sláma T, et al.: Severity of hearing loss after platinum chemotherapy in childhood cancer survivors. Pediatr Blood Cancer 69 (9): e29755, 2022. [PUBMED Abstract]
Beyea JA, Lau C, Cooke B, et al.: Long-Term Incidence and Predictors of Significant Hearing Loss Requiring Hearing Assistive Devices Among Childhood Cancer Survivors: A Population-Based Study. J Clin Oncol 38 (23): 2639-2646, 2020. [PUBMED Abstract]
Whelan K, Stratton K, Kawashima T, et al.: Auditory complications in childhood cancer survivors: a report from the childhood cancer survivor study. Pediatr Blood Cancer 57 (1): 126-34, 2011. [PUBMED Abstract]
Moke DJ, Luo C, Millstein J, et al.: Prevalence and risk factors for cisplatin-induced hearing loss in children, adolescents, and young adults: a multi-institutional North American cohort study. Lancet Child Adolesc Health 5 (4): 274-283, 2021. [PUBMED Abstract]
Clemens E, de Vries AC, Pluijm SF, et al.: Determinants of ototoxicity in 451 platinum-treated Dutch survivors of childhood cancer: A DCOG late-effects study. Eur J Cancer 69: 77-85, 2016. [PUBMED Abstract]
Brock PR, Knight KR, Freyer DR, et al.: Platinum-induced ototoxicity in children: a consensus review on mechanisms, predisposition, and protection, including a new International Society of Pediatric Oncology Boston ototoxicity scale. J Clin Oncol 30 (19): 2408-17, 2012. [PUBMED Abstract]
Hobbie WL, Li Y, Carlson C, et al.: Late effects in survivors of high-risk neuroblastoma following stem cell transplant with and without total body irradiation. Pediatr Blood Cancer 69 (3): e29537, 2022. [PUBMED Abstract]
Kushner BH, Budnick A, Kramer K, et al.: Ototoxicity from high-dose use of platinum compounds in patients with neuroblastoma. Cancer 107 (2): 417-22, 2006. [PUBMED Abstract]
Landier W, Knight K, Wong FL, et al.: Ototoxicity in children with high-risk neuroblastoma: prevalence, risk factors, and concordance of grading scales--a report from the Children's Oncology Group. J Clin Oncol 32 (6): 527-34, 2014. [PUBMED Abstract]
Lewis MJ, DuBois SG, Fligor B, et al.: Ototoxicity in children treated for osteosarcoma. Pediatr Blood Cancer 52 (3): 387-91, 2009. [PUBMED Abstract]
Freyer DR, Chen L, Krailo MD, et al.: Effects of sodium thiosulfate versus observation on development of cisplatin-induced hearing loss in children with cancer (ACCL0431): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol 18 (1): 63-74, 2017. [PUBMED Abstract]
Brock PR, Maibach R, Childs M, et al.: Sodium Thiosulfate for Protection from Cisplatin-Induced Hearing Loss. N Engl J Med 378 (25): 2376-2385, 2018. [PUBMED Abstract]
Orgel E, Knight KR, Villaluna D, et al.: Reevaluation of sodium thiosulfate otoprotection using the consensus International Society of Paediatric Oncology Ototoxicity Scale: A report from the Children's Oncology Group study ACCL0431. Pediatr Blood Cancer : e30550, 2023. [PUBMED Abstract]
Clemens E, de Vries AC, Am Zehnhoff-Dinnesen A, et al.: Hearing loss after platinum treatment is irreversible in noncranial irradiated childhood cancer survivors. Pediatr Hematol Oncol 34 (2): 120-129, 2017. [PUBMED Abstract]
Kolinsky DC, Hayashi SS, Karzon R, et al.: Late onset hearing loss: a significant complication of cancer survivors treated with Cisplatin containing chemotherapy regimens. J Pediatr Hematol Oncol 32 (2): 119-23, 2010. [PUBMED Abstract]
Fouladi M, Gururangan S, Moghrabi A, et al.: Carboplatin-based primary chemotherapy for infants and young children with CNS tumors. Cancer 115 (14): 3243-53, 2009. [PUBMED Abstract]
Jehanne M, Lumbroso-Le Rouic L, Savignoni A, et al.: Analysis of ototoxicity in young children receiving carboplatin in the context of conservative management of unilateral or bilateral retinoblastoma. Pediatr Blood Cancer 52 (5): 637-43, 2009. [PUBMED Abstract]
Qaddoumi I, Bass JK, Wu J, et al.: Carboplatin-associated ototoxicity in children with retinoblastoma. J Clin Oncol 30 (10): 1034-41, 2012. [PUBMED Abstract]
Funt SA, Knezevic A, Wilson K, et al.: Ototoxicity associated with high-dose carboplatin for patients with previously treated germ cell tumors. Cancer 129 (24): 3952-3961, 2023. [PUBMED Abstract]
Hua C, Bass JK, Khan R, et al.: Hearing loss after radiotherapy for pediatric brain tumors: effect of cochlear dose. Int J Radiat Oncol Biol Phys 72 (3): 892-9, 2008. [PUBMED Abstract]
Bhandare N, Jackson A, Eisbruch A, et al.: Radiation therapy and hearing loss. Int J Radiat Oncol Biol Phys 76 (3 Suppl): S50-7, 2010. [PUBMED Abstract]
Merchant TE, Gould CJ, Xiong X, et al.: Early neuro-otologic effects of three-dimensional irradiation in children with primary brain tumors. Int J Radiat Oncol Biol Phys 58 (4): 1194-207, 2004. [PUBMED Abstract]
Bass JK, Hua CH, Huang J, et al.: Hearing Loss in Patients Who Received Cranial Radiation Therapy for Childhood Cancer. J Clin Oncol 34 (11): 1248-55, 2016. [PUBMED Abstract]
Cheuk DK, Billups CA, Martin MG, et al.: Prognostic factors and long-term outcomes of childhood nasopharyngeal carcinoma. Cancer 117 (1): 197-206, 2011. [PUBMED Abstract]
Merchant TE, Hua CH, Shukla H, et al.: Proton versus photon radiotherapy for common pediatric brain tumors: comparison of models of dose characteristics and their relationship to cognitive function. Pediatr Blood Cancer 51 (1): 110-7, 2008. [PUBMED Abstract]
Paulino AC, Lobo M, Teh BS, et al.: Ototoxicity after intensity-modulated radiation therapy and cisplatin-based chemotherapy in children with medulloblastoma. Int J Radiat Oncol Biol Phys 78 (5): 1445-50, 2010. [PUBMED Abstract]
Trendowski MR, Baedke JL, Sapkota Y, et al.: Clinical and genetic risk factors for radiation-associated ototoxicity: A report from the Childhood Cancer Survivor Study and the St. Jude Lifetime Cohort. Cancer 127 (21): 4091-4102, 2021. [PUBMED Abstract]
Murphy B, Jackson A, Bass JK, et al.: Modeling the Risk of Hearing Loss From Radiation Therapy in Childhood Cancer Survivors: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 446-456, 2024. [PUBMED Abstract]
Chao C, Bhatia S, Xu L, et al.: Chronic Comorbidities Among Survivors of Adolescent and Young Adult Cancer. J Clin Oncol 38 (27): 3161-3174, 2020. [PUBMED Abstract]
Bass JK, Wang F, Thaxton ME, et al.: Association of hearing loss with patient-reported functional outcomes in adult survivors of childhood cancer. J Natl Cancer Inst 116 (4): 596-605, 2024. [PUBMED Abstract]
Gurney JG, Tersak JM, Ness KK, et al.: Hearing loss, quality of life, and academic problems in long-term neuroblastoma survivors: a report from the Children's Oncology Group. Pediatrics 120 (5): e1229-36, 2007. [PUBMED Abstract]
Olivier TW, Bass JK, Ashford JM, et al.: Cognitive Implications of Ototoxicity in Pediatric Patients With Embryonal Brain Tumors. J Clin Oncol 37 (18): 1566-1575, 2019. [PUBMED Abstract]
Brinkman TM, Bass JK, Li Z, et al.: Treatment-induced hearing loss and adult social outcomes in survivors of childhood CNS and non-CNS solid tumors: Results from the St. Jude Lifetime Cohort Study. Cancer 121 (22): 4053-61, 2015. [PUBMED Abstract]
Shields CL, Bas Z, Tadepalli S, et al.: Long-term (20-year) real-world outcomes of intravenous chemotherapy (chemoreduction) for retinoblastoma in 964 eyes of 554 patients at a single centre. Br J Ophthalmol 104 (11): 1548-1555, 2020. [PUBMED Abstract]
Francis JH, Brodie SE, Marr B, et al.: Efficacy and Toxicity of Intravitreous Chemotherapy for Retinoblastoma: Four-Year Experience. Ophthalmology 124 (4): 488-495, 2017. [PUBMED Abstract]
Shields CL, Shields JA, Cater J, et al.: Plaque radiotherapy for retinoblastoma: long-term tumor control and treatment complications in 208 tumors. Ophthalmology 108 (11): 2116-21, 2001. [PUBMED Abstract]
Schefler AC, Cicciarelli N, Feuer W, et al.: Macular retinoblastoma: evaluation of tumor control, local complications, and visual outcomes for eyes treated with chemotherapy and repetitive foveal laser ablation. Ophthalmology 114 (1): 162-9, 2007. [PUBMED Abstract]
Nguyen SM, Sison J, Jones M, et al.: Lens Dose-Response Prediction Modeling and Cataract Incidence in Patients With Retinoblastoma After Lens-Sparing or Whole-Eye Radiation Therapy. Int J Radiat Oncol Biol Phys 103 (5): 1143-1150, 2019. [PUBMED Abstract]
Fabian ID, Stacey AW, Naeem Z, et al.: Strabismus in retinoblastoma survivors with long-term follow-up. J AAPOS 22 (4): 276.e1-276.e7, 2018. [PUBMED Abstract]
Kaste SC, Chen G, Fontanesi J, et al.: Orbital development in long-term survivors of retinoblastoma. J Clin Oncol 15 (3): 1183-9, 1997. [PUBMED Abstract]
Peylan-Ramu N, Bin-Nun A, Skleir-Levy M, et al.: Orbital growth retardation in retinoblastoma survivors: work in progress. Med Pediatr Oncol 37 (5): 465-70, 2001. [PUBMED Abstract]
Banerjee SC, Pottenger E, Petriccione M, et al.: Impact of enucleation on adult retinoblastoma survivors' quality of life: A qualitative study of survivors' perspectives. Palliat Support Care 18 (3): 322-331, 2020. [PUBMED Abstract]
Shen CJ, Kry SF, Buchsbaum JC, et al.: Retinopathy, Optic Neuropathy, and Cataract in Childhood Cancer Survivors Treated With Radiation Therapy: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 431-445, 2024. [PUBMED Abstract]
Raney RB, Anderson JR, Kollath J, et al.: Late effects of therapy in 94 patients with localized rhabdomyosarcoma of the orbit: Report from the Intergroup Rhabdomyosarcoma Study (IRS)-III, 1984-1991. Med Pediatr Oncol 34 (6): 413-20, 2000. [PUBMED Abstract]
Paulino AC, Simon JH, Zhen W, et al.: Long-term effects in children treated with radiotherapy for head and neck rhabdomyosarcoma. Int J Radiat Oncol Biol Phys 48 (5): 1489-95, 2000. [PUBMED Abstract]
Hol MLF, Indelicato DJ, Rotondo RL, et al.: Dose-Effect Analysis of Early Changes in Orbital Bone Morphology After Radiation Therapy for Rhabdomyosarcoma. Pract Radiat Oncol 10 (1): 53-58, 2020. [PUBMED Abstract]
Hol MLF, Indelicato DJ, Slater O, et al.: Facial deformation following treatment for pediatric head and neck rhabdomyosarcoma; the difference between treatment modalities. Results of a trans-Atlantic, multicenter cross-sectional cohort study. Pediatr Blood Cancer 70 (8): e30412, 2023. [PUBMED Abstract]
Wan MJ, Ullrich NJ, Manley PE, et al.: Long-term visual outcomes of optic pathway gliomas in pediatric patients without neurofibromatosis type 1. J Neurooncol 129 (1): 173-8, 2016. [PUBMED Abstract]
Kelly JP, Leary S, Khanna P, et al.: Longitudinal measures of visual function, tumor volume, and prediction of visual outcomes after treatment of optic pathway gliomas. Ophthalmology 119 (6): 1231-7, 2012. [PUBMED Abstract]
Liu APY, Hastings C, Wu S, et al.: Treatment burden and long-term health deficits of patients with low-grade gliomas or glioneuronal tumors diagnosed during the first year of life. Cancer 125 (7): 1163-1175, 2019. [PUBMED Abstract]
de Blank PM, Fisher MJ, Lu L, et al.: Impact of vision loss among survivors of childhood central nervous system astroglial tumors. Cancer 122 (5): 730-9, 2016. [PUBMED Abstract]
Whelan KF, Stratton K, Kawashima T, et al.: Ocular late effects in childhood and adolescent cancer survivors: a report from the childhood cancer survivor study. Pediatr Blood Cancer 54 (1): 103-9, 2010. [PUBMED Abstract]
Chodick G, Sigurdson AJ, Kleinerman RA, et al.: The Risk of Cataract among Survivors of Childhood and Adolescent Cancer: A Report from the Childhood Cancer Survivor Study. Radiat Res 185 (4): 366-74, 2016. [PUBMED Abstract]
Alloin AL, Barlogis V, Auquier P, et al.: Prevalence and risk factors of cataract after chemotherapy with or without central nervous system irradiation for childhood acute lymphoblastic leukaemia: an LEA study. Br J Haematol 164 (1): 94-100, 2014. [PUBMED Abstract]
Bates JE, Indelicato DJ, Morris CG, et al.: Visual decline in pediatric survivors of brain tumors following radiotherapy. Acta Oncol 59 (10): 1257-1262, 2020. [PUBMED Abstract]
Hoehn ME, Vestal R, Calderwood J, et al.: Ocular Complications in School-Age Children and Adolescents after Allogeneic Bone Marrow Transplantation. Am J Ophthalmol 213: 153-160, 2020. [PUBMED Abstract]
Ferry C, Gemayel G, Rocha V, et al.: Long-term outcomes after allogeneic stem cell transplantation for children with hematological malignancies. Bone Marrow Transplant 40 (3): 219-24, 2007. [PUBMED Abstract]
Fahnehjelm KT, Törnquist AL, Olsson M, et al.: Visual outcome and cataract development after allogeneic stem-cell transplantation in children. Acta Ophthalmol Scand 85 (7): 724-33, 2007. [PUBMED Abstract]
Gurney JG, Ness KK, Rosenthal J, et al.: Visual, auditory, sensory, and motor impairments in long-term survivors of hematopoietic stem cell transplantation performed in childhood: results from the Bone Marrow Transplant Survivor study. Cancer 106 (6): 1402-8, 2006. [PUBMED Abstract]
Kal HB, VAN Kempen-Harteveld ML: Induction of severe cataract and late renal dysfunction following total body irradiation: dose-effect relationships. Anticancer Res 29 (8): 3305-9, 2009. [PUBMED Abstract]
Holmström G, Borgström B, Calissendorff B: Cataract in children after bone marrow transplantation: relation to conditioning regimen. Acta Ophthalmol Scand 80 (2): 211-5, 2002. [PUBMED Abstract]
Horwitz M, Auquier P, Barlogis V, et al.: Incidence and risk factors for cataract after haematopoietic stem cell transplantation for childhood leukaemia: an LEA study. Br J Haematol 168 (4): 518-25, 2015. [PUBMED Abstract]
Jeppesen H, Kielsen K, Siersma V, et al.: Ocular graft-versus-host disease and dry eye disease after paediatric haematopoietic stem cell transplantation - incidence and risk factors. Bone Marrow Transplant 57 (3): 487-498, 2022. [PUBMED Abstract]
Efectos tardíos en el aparato urinario
La toxicidad aguda que produce el tratamiento del cáncer en el aparato urinario es bien conocida. Se sabe menos acerca de los desenlaces genitourinarios en los sobrevivientes a largo plazo.[1] La evidencia de lesión renal a largo plazo en los sobrevivientes de cáncer infantil es limitada porque los estudios se caracterizan por el tamaño pequeño de la muestra, el sesgo de selección de la cohorte y el sesgo de participación, la evaluación transversal, la heterogeneidad en el tiempo desde el tratamiento y el método de confirmación. En particular, se deben considerar las imprecisiones en el diagnóstico de la disfunción renal crónica mediante el cálculo de las ecuaciones de disfunción glomerular.[2]
Se evaluó la incidencia de intervenciones quirúrgicas importantes y tardías entre los sobrevivientes de cáncer infantil a través de los datos del Childhood Cancer Survivor Study (CCSS). Los sobrevivientes de cáncer infantil tenían más probabilidad de someterse a una intervención quirúrgica importante tardía renal o urinaria que sus hermanos de ambos sexos (riesgo relativo [RR] ajustado, 2,0). La cirugía locorregional o los tratamientos oncológicos con radioterapia se relacionaron con someterse a una intervención quirúrgica tardía en la misma región corporal o sistema orgánico.[3]
Los tratamientos para el cáncer que predisponen a lesiones renales o hipertensión más tarde en la vida son los siguientes:
El riesgo y el grado de la disfunción renal dependen del tipo e intensidad del tratamiento; la interpretación de los estudios se ve comprometida por la variabilidad de las pruebas utilizadas.
La incidencia y los factores de riesgo de la insuficiencia renal de inicio tardío entre los sobrevivientes a largo plazo de cáncer infantil no han sido bien estudiados.
Entre los 25 530 participantes del CCSS (mediana de seguimiento, 22,3 años), la incidencia acumulada a 35 años de insuficiencia renal de inicio tardío autonotificada, definida como diálisis, trasplante renal o muerte atribuible a la enfermedad renal, fue del 1,7 % (en comparación con el 0,2 % en la cohorte de hermanos).[4]
Los factores de riesgo independientes del tratamiento para la insuficiencia renal de inicio tardío incluían dosis altas (>15 Gy) de radiación que afectaba el riñón (RR, 4,0), dosis altas (>250 mg/m2) de antraciclina (RR, 1,6), cualquier dosis de ifosfamida (RR, 2,6) y nefrectomía (RR, 1,9).
Los factores de riesgo modificables asociados a la insuficiencia renal de inicio tardío fueron la hipertensión (RR, 14,4 con antecedentes de nefrectomía; RR, 5,9 sin antecedentes de nefrectomía) y la diabetes (RR, 2,2).
Son pocos los estudios de gran escala en los que se evalúan los desenlaces tardíos para la salud renal y los factores de riesgo para la disfunción renal en sobrevivientes tratados con modalidades potencialmente nefrotóxicas.
Evidencia (disfunción renal en sobrevivientes de cáncer infantil):
En el análisis renal del Dutch Childhood Cancer Survivor Study (DCCSS)-LATER 2, un estudio de cohortes transversal de ámbito nacional, se evaluó a 1024 sobrevivientes de cáncer (mayores de 18 años en el momento del estudio). Habían pasado 5 años o más desde el diagnóstico y los pacientes recibieron tratamiento entre 1963 y 2001 con nefrectomía, radioterapia abdominal, irradiación total del cuerpo (ITC), cisplatino, carboplatino, ifosfamida, dosis altas de ciclofosfamida o trasplante de células madre hematopoyéticas (TCMH). Estos sobrevivientes se compararon con 500 controles emparejados por edad y sexo.[5]
En el 3,7 % de los sobrevivientes (mediana de edad, 32 años), la tasa de filtración glomerular (TFG) fue inferior a 60 ml/min/1,73 m2, en comparación con 0 % en el grupo de control, dado que ninguno de los participantes de este grupo mostraron una disminución de la TFG por debajo de ese límite.
La insuficiencia renal terminal estuvo presente en el 1,1 % de los sobrevivientes.
La enfermedad renal crónica, según los umbrales de edad (TFG inferior a 75 para las personas de <40 años, TFG inferior a 60 para las personas de 40–65 años, y TFG inferior a 40 para las personas de <65 años), fue del 6,6 % en los sobrevivientes, frente al 0,2 % en los controles.
Los factores de riesgo de enfermedad renal crónica, basados en análisis multivariantes, incluían el tratamiento con nefrectomía (oportunidad relativa [OR], 3,7), radioterapia abdominal (OR, 1,8), ifosfamida (especialmente >12 gm/m2; OR, 2,9) y dosis de cisplatino superiores a 500 mg/m2 (OR, 7,2).
Se identificó albuminuria (relación albúmina/creatinina superior a 3 mg/mmol) en el 16,2 % de los sobrevivientes y en el 1,2 % de los controles. Los factores de riesgo de albuminuria incluyeron el tratamiento con ICT (OR, 2,3), la radioterapia abdominal superior a 30 Gy (OR, 2,6) y la ifosfamida (OR, 1,6).
Tras una mediana de seguimiento de 25,5 años, la prevalencia general de pérdidas electrolíticas en los sobrevivientes de cáncer infantil (magnesio 5,6 %, potasio 4,5 %, fosfato 5,5 %) no fue superior a la de los controles.[6]
Los factores de riesgo de hipertensión fueron la radioterapia abdominal y la ICT. La hipertensión se relaciona con una disminución de la TFG en los sobrevivientes de cáncer infantil.[6]
Los investigadores franceses del CCSS vincularon datos del sistema nacional de salud con datos clínicos y de tratamiento procedentes de historias clínicas para evaluar el riesgo a largo plazo de hospitalización relacionada con los riñones o el aparato urinario en sobrevivientes de cáncer infantil.[7]
De los 5498 sobrevivientes (mediana de edad en el momento del diagnóstico, 5 años; mediana de tiempo transcurrido desde el diagnóstico, 22,9 años), 230 fueron hospitalizados por indicaciones renales o de las vías urinarias.
La indicación más frecuente de hospitalización renal fue la diálisis (64,5 %), mientras que las indicaciones de hospitalización relacionada con el aparato urinario incluyeron la urolitiasis (73,2 %), seguida de las enfermedades de la vejiga (15,7 %).
El riesgo de hospitalización renal casi se triplicó (razón de hospitalización estandarizada, 2,9; intervalo de confianza [IC] del 95 %, 2,4–3,4), en comparación con la población general.
La exposición de más del 10 % del volumen renal a, al menos, 20 Gy de radiación se asoció con un riesgo 2 veces mayor (razón de hospitalización estandarizada, 2,2; IC 95 %, 1,3–3,6) de hospitalización por causas renales.
Los sobrevivientes tratados con nefrectomía y dosis altas de ifosfamida (>60 g/m2) tenían un riesgo más de 20 veces mayor de hospitalización por causas renales (RR, 23,7; IC 95 %, 5,1–110,0).
Se evaluaron las comorbilidades crónicas en una cohorte de 6778 sobrevivientes de 2 años de cáncer en adolescentes y adultos jóvenes (AAJ) (definidos como personas diagnosticadas de cáncer entre los 15 y los 39 años de edad), con una mediana de seguimiento de 5,1 años tras el diagnóstico de cáncer. Los sobrevivientes de cáncer AAJ presentaban un riesgo entre 2 y 3 veces mayor de insuficiencia renal. El riesgo de presentar comorbilidades crónicas se comparó con el de personas emparejadas sin antecedentes de cáncer.[8]
Factores relacionados con el tratamiento que afectan el riñón
Los tratamientos del cáncer que predisponen a lesión renal e hipertensión tardías son los siguientes:[9,10]
Nefrectomía
Los sobrevivientes de cáncer infantil sometidos a nefrectomía tienen riesgo de lesión por hiperfiltración.
Por lo general, la hipertrofia compensadora del riñón contralateral se produce después de la nefrectomía pero, con el tiempo, el daño renal quizás se manifieste como una reducción de filtración glomerular, microalbuminuria y proteinuria, hipertensión y, con poca frecuencia, glomeruloesclerosis focal que conduce a insuficiencia renal crónica.[9,11]
Los pacientes con tumor de Wilms unilateral no sindrómico tratados con nefrectomía radical unilateral tienen una incidencia acumulada a los 35 años de insuficiencia renal tardía del 2,4 %, una tasa aproximadamente 10 veces mayor en relación con los hermanos. Ninguna exposición relacionada con la quimioterapia o la radioterapia se asoció con insuficiencia renal tardía en un estudio del CCSS sobre resultados tardíos de salud en sobrevivientes de tumores de Wilms (n = 2008).[12]
En un metanálisis se examinaron los datos de 23 estudios de pacientes con tumor de Wilms (293 casos unilaterales y 386 casos bilaterales). En el estudio se demostró un aumento significativo de las probabilidades de presentar una función renal anormal (OR, 4,29; IC 95 %, 1,02–18,00) y de tener una TFG estimada más baja (media, -8,99) entre los pacientes que se trataron con nefrectomía radical, en comparación con los tratados con cirugía con conservación de nefronas. Los pacientes con tumor de Wilms bilateral (media de seguimiento, 7,7 años) tratados con nefrectomía radical y cirugía conservadora de nefronas contralateral tenían mayores probabilidades de presentar una función renal anormal (OR, 3,82; IC 95 %, 1,76–8,33) e hipertensión (OR, 5,81; IC 95 %, 1,31–25,86), en comparación con los tratados con cirugía conservadora de nefronas bilateral.[13]
Quimioterapia
La administración de cisplatino, carboplatino e ifosfamida son factores de riesgo para el desarrollo de disfunción tubular renal.[5]
Cisplatino
Es posible que el cisplatino cause daño glomerular y tubular que conduce a una disminución de la TFG y pérdida de electrólitos (en particular, magnesio, calcio y potasio).
Se notificó nefrotoxicidad relacionada con el cisplatino en el 30 % al 100 % de los niños expuestos.[14] No obstante, la prevalencia de disfunción renal persistente en sobrevivientes a largo plazo es considerablemente más baja.
En el análisis renal del DCCSS-LATER 2, un estudio de cohortes transversal de ámbito nacional, se evaluó a 1024 sobrevivientes (de 18 años o más en el momento del estudio) en busca de disfunción tubular a largo plazo. Habían pasado 5 años o más desde el diagnóstico y los pacientes recibieron tratamiento entre 1963 y 2001 con nefrectomía, radioterapia abdominal, ITC, cisplatino, carboplatino, ifosfamida, dosis altas de ciclofosfamida o TCMH. Los sobrevivientes se compararon con 500 controles emparejados por edad y sexo.[6]
En general, la prevalencia y las OR de la pérdida de magnesio aumentaron de manera significativa en los sobrevivientes de cáncer infantil tratados con cisplatino solo (25,8 %) o en combinación con carboplatino (23,1 %).
La pérdida de magnesio se asoció con todas las dosis de cisplatino, pero fue mayor para una dosis acumulada superior a 500 mg/m2 (OR, 22,0). Los sobrevivientes de cáncer infantil expuestos solo a cisplatino presentaban riesgo de pérdida de potasio en comparación con los controles (prevalencia, 12 %; OR, 3,2).
A dosis acumuladas de cisplatino superiores a 500 mg/m2, aumentaron las probabilidades de pérdida de potasio (OR, 17,7) y fosfato (OR, 3,6).
En 63 niños tratados con derivados del platino, la TFG fue inferior a 60 ml/min/1,73 m2 en el 11 % de los niños, y la hipomagnesemia que requería suplementos orales se presentó en el 7 % de los niños a los 10 años de finalizar el tratamiento.[15]
Carboplatino
El carboplatino es un análogo del cisplatino y es menos nefrotóxico que el cisplatino.
En un estudio prospectivo longitudinal de cohortes realizado en un solo centro, de niños controlados durante más de 10 años después del tratamiento con cisplatino o carboplatino, se encontró que una edad mayor en el momento del tratamiento fue el factor de riesgo más importante de nefrotoxicidad, en especial, en los pacientes que recibieron carboplatino, mientras que el cronograma de dosis de cisplatino y las dosis acumuladas de carboplatino también fueron factores de predicción importantes de toxicidad. La nefrotoxicidad de los derivados del platino no cambió significativamente durante 10 años.[15]
En el análisis renal del DCCSS-LATER 2, un estudio de cohortes transversal de ámbito nacional, se evaluó a 1024 sobrevivientes (de 18 años o más en el momento del estudio) en busca de disfunción tubular a largo plazo. En el análisis multivariante de riesgos, tras una mediana de seguimiento de 25,5 años, el carboplatino se asoció con la pérdida de potasio a una dosis acumulada superior a 2800 mg/m2 (OR, 5,1).[6]
La combinación de carboplatino e ifosfamida quizás se vincule con más daño renal que la combinación de cisplatino e ifosfamida.[15-17]
Ifosfamida
La ifosfamida también causa toxicidad glomerular y tubular, con acidosis tubular renal y síndrome de Fanconi, que es un defecto tubular proximal caracterizado por deterioro de la reabsorción de glucosa, aminoácidos, fosfato y bicarbonato.
En el análisis renal DCCSS-LATER 2, un estudio de cohortes transversal de ámbito nacional, se evaluó a 1024 sobrevivientes (de 18 años o más en el momento del estudio) que habían recibido tratamiento potencialmente nefrotóxico para la disfunción tubular a largo plazo. En el análisis multivariante de riesgos, tras una mediana de seguimiento de 25,5. años, la ifosfamida se asoció con pérdida de potasio (OR, 2,4) y pérdida de fosfato (OR, 2,3) (con dosis acumulada >42 g/m2).[6]
La ifosfamida en dosis superiores a 60 g/m2, edad menor de 5 años en el momento del tratamiento, y la combinación de cisplatino con carboplatino aumentan el riesgo de toxicidad tubular renal relacionada con la ifosfamida.[18-20]
Metotrexato
Se notificó que las dosis altas de metotrexato (1000–33 000 mg/m2) causan disfunción renal aguda en hasta 12,4 % de los pacientes. No se han descrito las secuelas renales a largo plazo.[21]
Radioterapia
La radioterapia dirigida al riñón en ocasiones causa nefritis por radiación o nefropatía después de un período de latencia de 3 a 12 meses.
El riñón es relativamente radiosensible, con una dosis de tolerancia de 20 Gy.[22]
Se considera poco probable que las dosis de 18 Gy causen secuelas renales graves o crónicas. En cambio, hasta el 50 % de aquellos tratados con 20 Gy presentan disfunción glomerular o hipertensión en el transcurso de 20 años.[23]
Los datos cuantitativos específicos son escasos pero, en un estudio de 108 niños tratados por tumor de Wilms sometidos a nefrectomía unilateral, se observó que el 41 % de los niños que recibieron menos de 12 Gy dirigidos al riñón contralateral restante, el 56 % de los niños que recibieron de 12 a 24 Gy y el 91 % de los niños que recibieron más de 24 Gy sufrieron una disminución de la depuración de creatinina (TFG, <63 ml/min/m2).[24]
En un informe del German Registry for the Evaluation of Side Effects after Radiation in Childhood and Adolescence (consorcio RISK), se indica que se evaluó a 126 pacientes que recibieron radioterapia dirigida a partes de los riñones por distintos cánceres. Todos los pacientes también habían recibido quimioterapia posiblemente nefrotóxica. Los volúmenes renales completos expuestos a radiación de más de 20 Gy (P = 031) o 30 Gy (P = 0,003) de radiación se relacionaron con un mayor riesgo de nefrotoxicidad.[25]
Los factores de riesgo de lesiones por radiación son los siguientes:
Edad en el momento de la radioterapia.
Los neonatos tienen mayor sensibilidad a la radioterapia; las dosis de 12 Gy a 24 Gy en fracciones de 1,25 Gy a 1,5 Gy dirigidas a todo el riñón se relacionaron con una disminución de la TFG.[26]
Sin embargo, para los niños mayores, no hay evidencia convincente de que la edad en el momento de la radioterapia esté relacionada con una lesión renal.[26]
Radioterapia unilateral versus bilateral.
En la experiencia del National Wilms Tumor Study, la insuficiencia renal fue más común en niños con tumores bilaterales que en niños con tumores unilaterales.[27]
Los efectos de la radiación también dependen de que se haya administrado radioterapia a parte o a todo el riñón. La insuficiencia renal es poco frecuente después de la administración de un volumen parcial de radiación en dosis entre 12 Gy y 27 Gy.[28]
Cuando no se usan ciertos fármacos como la ciclosporina y el tenipósido, las (ICT) de hasta 13 Gy se relacionan con una incidencia de toxicidad renal de menos del 8 %.[29]
Trasplante de células madre hematopoyéticas.
La nefropatía crónica es una complicación a largo plazo de un trasplante de células madre hematopoyéticas (TCMH) que se relacionó de modo variable con lesión renal aguda, funcionamiento renal más bajo antes del trasplante, ICT, regímenes de acondicionamiento como aquellos con fludarabina, enfermedad de injerto contra huésped y administración de inhibidores de la calcineurina.[30-32]
La mayoría de los informes sobre desenlaces renales en sobrevivientes a largo plazo de cáncer infantil tratados con TCMH se limitan a desenlaces descriptivos de cohortes muy pequeñas.
Para obtener más información, consultar la sección Efectos tardíos en el aparato urinario en Complicaciones del trasplante de células madre hematopoyéticas en pediatría, enfermedad de injerto contra huésped y efectos tardíos.
En una revisión exhaustiva del Pediatric Normal Tissue Effects in the Clinic (PENTEC) realizada por el grupo de trabajo genitourinario se evaluaron los factores dosimétricos y clínicos de las relaciones dosis-volumen de radiación para la disfunción genitourinaria, incluso riñón, vejiga e hipertensión, para neoplasias malignas pediátricas. También se tuvo en cuenta el efecto de la quimioterapia.[33]
El riesgo de toxicidad de la irradiación de abdomen completo con 10,5 Gy fue bajo entre los pacientes con tumor de Wilms (riesgos de toxicidad ≥grado 2, 4 % y riesgos de toxicidad ≥grado 3, 1 %).
Para la ICT fraccionada de 12 Gy con varias combinaciones de quimioterapia, el riesgo de toxicidad grave fue bajo, pero la toxicidad de grado bajo fue más común (riesgos de toxicidad ≥grado 2, 8 % y riesgos de toxicidad ≥grado 3, 3 %).
Con una dosis de cisplatino de 480 mg/m2, se produjo un riesgo del 3 % de toxicidad de grado 3 o superior sin radiación, y un riesgo del 5 % cuando el 26 % del riñón recibió 10 Gy o más.
Con una dosis de ifosfamida de 63 g/m2, se produjo un riesgo del 5 % de toxicidad de grado 3 o superior sin radiación, y un riesgo de toxicidad del 10 % cuando el 42 % del riñón recibió 10 Gy o más.
Funcionamiento renal en sobrevivientes de tumor de Wilms no sindrómico
Los investigadores utilizaron el CCSS para evaluar la mortalidad a largo plazo y las afecciones crónicas entre los sobrevivientes de tumores de Wilms unilaterales no sindrómicos (n = 2008) diagnosticados a lo largo de 3 décadas. La incidencia acumulada a 35 años de insuficiencia renal tardía entre los sobrevivientes fue del 2,4 % (tasa aproximadamente 10 veces mayor en relación con los hermanos). La tasa de insuficiencia renal tardía a 20 años fue del 1,7 % y aumentó con la edad. Los sobrevivientes presentaban un mayor riesgo de insuficiencia renal (RR, 11,9), en comparación con los hermanos.[34]
Los investigadores del estudio St. Jude Life (SJLIFE) evaluaron las consecuencias de modalidades específicas de tratamiento sobre el funcionamiento renal a largo plazo y la presión arterial en adultos sobrevivientes de tumor de Wilms.
En 40 sobrevivientes de tumor de Wilms no sindrómico unilateral, la prevalencia de hipertensión aumentó significativamente en los sobrevivientes que no se irradiaron (25 %) y en los que se irradiaron (35 %) en comparación con la prevalencia en 35 controles sin cáncer (0 %).
Ningún participante irradiado o no irradiado presentó una TFG estimada (creatinina y cistatina C) inferior a 60 ml/min/1,73 m2.
Los adultos sobrevivientes de un tumor de Wilms unilateral, no metastásico y no sindrómico, tratados con radioterapia dirigida a todo el abdomen, presentan riesgo de deterioro del funcionamiento renal. En un estudio piloto de la cohorte del SJLIFE, se evaluó la exactitud de las ecuaciones que evalúan el funcionamiento renal en este grupo de pacientes.[35]
Es posible que el funcionamiento renal disminuya en las sobrevivientes mujeres (n = 48) que recibieron radioterapia dirigida a todo el abdomen en comparación con las sobrevivientes mujeres que no recibieron radioterapia dirigida a todo el abdomen y con los controles.
El funcionamiento renal de los hombres que recibieron radioterapia dirigida a todo el abdomen (n = 27) no se vio alterado.
Los resultados de las ecuaciones de estimación no se correlacionaron con el aclaramiento plasmático de ácido tecnecioTc 99m-dietilentriamina pentacético (99mTc-DTPA) en las mujeres que recibieron irradiación ni en aquellas que no la recibieron.
La tasa estimada de TFG (creatinina) y la tasa estimada de TFG (creatinina y cistatina C) se correlacionaron con el aclaramiento plasmático de 99mTc-DTPA en los hombres que recibieron irradiación y en aquellos que no la recibieron.
En este informe se indica que las sobrevivientes mujeres que se sometieron a nefrectomía unilateral y se examinaron mediante la estimación de la tasa de TFG quizás se hayan clasificado de forma errónea con un funcionamiento renal normal. Sin embargo, se deben evaluar cohortes más numerosas de sobrevivientes de tumor de Wilms para validar estos hallazgos.
Factores genéticos que predisponen a disfunción renal
Muchos sobrevivientes de tumor de Wilms infantil que presentan insuficiencia renal crónica tienen síndromes que acompañan las variantes o deleciones del gen WT1 que predisponen a la enfermedad renal.
Los datos del National Wilms Tumor Study Group y el U.S. Renal Data System indican que la incidencia acumulada a 20 años de enfermedad renal en estadio terminal es del 74 % en niños con tumor de Wilms unilateral y síndrome de Denys-Drash; del 36 % en aquellos con síndrome WAGR (tumor de Wilms, aniridia, anormalidades genitourinarias y una variedad de alteraciones en el desarrollo neurológico), del 7 % para los pacientes varones con anomalías genitourinarias y del 0,6 % para los pacientes sin ninguna de estas afecciones.[36]
En los pacientes con tumores de Wilms bilaterales, la incidencia de enfermedad renal en estadio terminal es del 50 % en pacientes con síndrome de Denys-Drash, del 90 % en pacientes con síndrome WAGR, del 25 % en pacientes con anomalías genitourinarias y del 12 % en el resto de pacientes.[36,37]
La nefropatía en estadio terminal en pacientes con síndrome WAGR y anomalías genitourinarias tiende a presentarse relativamente tarde y, a menudo, durante la adolescencia o después de esta.[36]
Predicción del riesgo de insuficiencia renal
Los investigadores del CCSS presentaron modelos de predicción de insuficiencia renal usando características demográficas y de tratamiento de sobrevivientes a 5 años de cáncer infantil. Los modelos se validaron mediante datos de seguimiento de sobrevivientes del estudio SJLIFE y del National Wilms Tumor Study.[38]
En el CCSS, los antecedentes de insuficiencia renal posterior (es decir, diálisis, trasplante de riñón o muerte relacionada con el riñón) declarados por los sobrevivientes a los 40 años se confirmaron mediante la vinculación con la Organ Procurement and Transplantation Network and the National Death Index.
Los modelos integraron datos clínicos como la raza y la etnia, la edad en el momento del diagnóstico, la nefrectomía, la quimioterapia, la radioterapia, las anomalías genitourinarias congénitas y la hipertensión de inicio precoz para estimar las relaciones entre los posibles factores predictivos y la insuficiencia renal y la magnitud del riesgo.
Los modelos de predicción del riesgo identifican con precisión a los sobrevivientes con riesgo bajo, moderado y alto de insuficiencia renal tardía que podrían beneficiarse de la detección y la intervención.
La calculadora en línea en inglés se encuentra en el sitio web del CCSS.
Complicaciones vesicales relacionadas con el tratamiento
La cirugía pélvica o del sistema nervioso central, la quimioterapia con alquilantes, como ciclofosfamida o ifosfamida, la radioterapia pélvica y ciertos procedimientos quirúrgicos raquídeos y genitourinarios se relacionaron con los siguientes efectos tardíos en la vejiga urinaria:
Quimioterapia
Los alquilantes de tipo oxazafosforinas (ciclofosfamida e ifosfamida) y la exposición de la vejiga a la radioterapia se vincularon con la presentación de cistitis hemorrágica.
La cistitis hemorrágica relacionada con la quimioterapia se presenta como un efecto tóxico agudo y parece ser un efecto persistente poco frecuente en las cohortes de sobrevivientes a largo plazo clínicamente bien caracterizadas.[39,40]
En un estudio de 6119 niños tratados entre 1986 y 2010, (media de edad, 12,2 años ± 6,3 de desviación estándar), el 1,6 % de los pacientes (n = 97) presentaron cistitis hemorrágica (que se manifestó tras una media de 2,7 meses después de la terapia de inducción para el trasplante y una media de 12,4 meses después de la radiación pélvica), la mayoría (75 %) con puntuaciones de gravedad de II o III (escala, I–IV).[41]
Se excluyó del estudio a los pacientes con manifestaciones radiológicas de cálculos renales o vesicales, o de tumores que invadieron la pared de la vejiga.
La edad avanzada, el antecedente de un trasplante de médula ósea o de TCMH periféricas en sangre y la presencia del virus BK en la orina fueron factores de riesgo para la cistitis hemorrágica y se relacionaron con puntuaciones más altas de gravedad.
La exposición previa a la ciclofosfamida se relacionó con riesgo de carcinoma de vejiga. También se ha observado un exceso de prevalencia de tumores de vejiga en los sobrevivientes de tipos específicos de diagnóstico (por ejemplo, retinoblastoma hereditario) que apoyan la contribución de los factores genéticos en la presentación de neoplasias subsiguientes.[42,43]
Radioterapia
La radioterapia pélvica también se relaciona con un aumento de riesgo de cistitis hemorrágica que tal vez se presente de forma aguda o crónica.
El riesgo de cistitis hemorrágica inducida por la radiación es mayor en sobrevivientes tratados con dosis de radiación de más de 30 Gy dirigidos a toda la vejiga o más de 60 Gy dirigidos a una parte de la vejiga.[44]
La fibrosis y la contractura vesical a largo plazo es posible que se presenten como secuelas de la cistitis hemorrágica o la radioterapia.[44]
Cirugía
Los procedimientos quirúrgicos que comprometen el aparato genitourinario inferior tienen el potencial de alterar el funcionamiento normal de la vejiga y los mecanismos normales de micción.
Cualquier tratamiento del cáncer o infiltración tumoral que interrumpe la inervación de la vejiga puede tener efectos dañinos sobre el funcionamiento vesical que a veces se manifiestan como deterioro del almacenamiento de la vejiga, incapacidad para vaciar la vejiga o incontinencia.
Los niños sometidos a enterocistoplastía ileal para aumentar el volumen de la vejiga están en riesgo de presentar deficiencia de vitamina B12. Las concentraciones séricas de vitamina B12 disminuyen con el tiempo después del procedimiento; el mayor riesgo se presenta 7 años después de la operación.[45]
Trasplante de riñón
En un estudio de trasplantes de vísceras macizas con 13 318 sobrevivientes del Childhood Cancer Survivor Study (CCSS), 71 sobrevivientes tenían enfermedad renal en etapa terminal que justificaba un trasplante renal y 50 de ellos recibieron un trasplante de riñón.[46]
A los 35 años del diagnóstico de cáncer, la incidencia acumulada del trasplante de riñón fue del 0,39 %, y la incidencia acumulada de entrar en la lista de espera o de recibir un riñón fue del 0,54 %.
La exposición a la ifosfamida y el antecedente de recibir ICT se relacionaron con el cociente de riesgos instantáneos más alto de entrar en la lista de espera o recibir un trasplante de riñón.
La tasa de supervivencia a 5 años del trasplante de riñón fue del 93,5 %, que es similar a la de la población general en el mismo intervalo de edad.
En el Cuadro 21 se resumen los efectos tardíos renales y vesicales, y los exámenes médicos de detección correspondientes.
Cuadro 21. Efectos tardíos renales y vesicalesa
Tratamiento predisponente
Efectos renales o genitourinarios
Exámenes médicos de detección
NUS = nitrógeno ureico en sangre; AINE = antiinflamatorios no esteroideos; GR/CGA = glóbulos rojos por campo de gran aumento (examen microscópico).
Concentraciones de NUS, creatinina, Na, K, Cl, CO2, Ca, Mg, PO4
Análisis de orina
Hablar sobre deportes de contacto, seguridad cuando se practica ciclismo (por ejemplo, evitar las lesiones que causan los manubrios) y uso correcto de cinturones de seguridad (por ejemplo, usar los cinturones de seguridad subabdominales alrededor de la cadera, no alrededor de la cintura)
Aconsejar el uso cuidadoso de los AINE
Consulta nefrológica para pacientes con hipertensión, proteinuria o insuficiencia renal progresiva
Nefrectomía, cirugía pélvica, cistectomía
Hidrocele
Examen de los testículos
Cistectomía
Complicaciones relacionadas con la cistectomía (infecciones crónicas del aparato urinario, disfunción renal, reflujo vesicoureteral, hidronefrosis, depósito de cálculos, perforación espontánea de la neovejiga, deficiencia de vitamina 12, folato o caroteno [pacientes con enterocistoplastia ileal sola])
Evaluación urológica
Concentración de vitamina B12
Cirugía pélvica, cistectomía
Incontinencia urinaria, obstrucción de las vías urinarias
Antecedentes: hematuria, urgencia o frecuencia urinarias, incontinencia o retención urinaria, disuria, nocturia, flujo urinario anormal
Orientación sobre ingesta adecuada de líquidos, micción regular, búsqueda de atención médica por síntomas de disfunción miccional o infecciones de las vías urinarias, cumplimiento con el régimen de cateterismo vesical recomendado
Consulta urológica para pacientes con micción disfuncional o infecciones recidivantes de las vías urinarias
Ciclofosfamida o ifosfamida, radioterapia con exposición de la vejiga o las vías urinarias
Toxicidad vesical (cistitis hemorrágica, fibrosis de la vejiga, micción disfuncional, reflujo vesicoureteral, hidronefrosis)
Antecedentes: hematuria, urgencia o frecuencia urinarias, incontinencia o retención urinaria, disuria, nicturia, flujo urinario anormal
Análisis de orina
Cultivo de orina, proporción de calcio o creatinina en una muestra puntual de orina, y ecografía de los riñones y la vejiga para pacientes con hematuria microscópica (definida como ≥5 GR/CGA en por lo menos 2 ocasiones)
Derivación a nefrología o urología para pacientes con hematuria microscópica con cultivo negativo Y ecografía anormal, o proporción anormal de calcio o creatinina
Derivación a urología para pacientes con hematuria macroscópica y cultivo negativo
Shnorhavorian M, Friedman DL, Koyle MA: Genitourinary long-term outcomes for childhood cancer survivors. Curr Urol Rep 10 (2): 134-7, 2009. [PUBMED Abstract]
Green DM: Evaluation of renal function after successful treatment for unilateral, non-syndromic Wilms tumor. Pediatr Blood Cancer 60 (12): 1929-35, 2013. [PUBMED Abstract]
Dieffenbach BV, Murphy AJ, Liu Q, et al.: Cumulative burden of late, major surgical intervention in survivors of childhood cancer: a report from the Childhood Cancer Survivor Study (CCSS) cohort. Lancet Oncol 24 (6): 691-700, 2023. [PUBMED Abstract]
Dieffenbach BV, Liu Q, Murphy AJ, et al.: Late-onset kidney failure in survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. Eur J Cancer 155: 216-226, 2021. [PUBMED Abstract]
Kooijmans ECM, van der Pal HJH, Pluijm SMF, et al.: The Dutch Childhood Cancer Survivor Study (DCCSS)-LATER 2 kidney analysis examined long-term glomerular dysfunction in childhood cancer survivors. Kidney Int 102 (5): 1136-1146, 2022. [PUBMED Abstract]
Kooijmans ECM, van der Pal HJH, Pluijm SMF, et al.: Long-Term Tubular Dysfunction in Childhood Cancer Survivors; DCCSS-LATER 2 Renal Study. Cancers (Basel) 14 (11): , 2022. [PUBMED Abstract]
Mansouri I, Schwartz B, Vu-Bezin G, et al.: Risk of Renal or Urinary Related Hospitalization in Survivors of Childhood Cancer: Results from the French Childhood Cancer Survivor Study. Cancer Epidemiol Biomarkers Prev 32 (4): 572-581, 2023. [PUBMED Abstract]
Chao C, Bhatia S, Xu L, et al.: Chronic Comorbidities Among Survivors of Adolescent and Young Adult Cancer. J Clin Oncol 38 (27): 3161-3174, 2020. [PUBMED Abstract]
Dekkers IA, Blijdorp K, Cransberg K, et al.: Long-term nephrotoxicity in adult survivors of childhood cancer. Clin J Am Soc Nephrol 8 (6): 922-9, 2013. [PUBMED Abstract]
Mulder RL, Knijnenburg SL, Geskus RB, et al.: Glomerular function time trends in long-term survivors of childhood cancer: a longitudinal study. Cancer Epidemiol Biomarkers Prev 22 (10): 1736-46, 2013. [PUBMED Abstract]
Knijnenburg SL, Jaspers MW, van der Pal HJ, et al.: Renal dysfunction and elevated blood pressure in long-term childhood cancer survivors. Clin J Am Soc Nephrol 7 (9): 1416-27, 2012. [PUBMED Abstract]
Weil BR, Murphy AJ, Liu Q, et al.: Late Health Outcomes Among Survivors of Wilms Tumor Diagnosed Over Three Decades: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 41 (14): 2638-2650, 2023. [PUBMED Abstract]
Khondker A, Jain A, Groff ML, et al.: Late Kidney Effects of Nephron-Sparing vs Radical Nephrectomy for Wilms Tumor: A Systematic Review and Meta-Analysis. J Urol 207 (3): 513-523, 2022. [PUBMED Abstract]
Skinner R, Kaplan R, Nathan PC: Renal and pulmonary late effects of cancer therapy. Semin Oncol 40 (6): 757-73, 2013. [PUBMED Abstract]
Skinner R, Parry A, Price L, et al.: Persistent nephrotoxicity during 10-year follow-up after cisplatin or carboplatin treatment in childhood: relevance of age and dose as risk factors. Eur J Cancer 45 (18): 3213-9, 2009. [PUBMED Abstract]
Marina NM, Poquette CA, Cain AM, et al.: Comparative renal tubular toxicity of chemotherapy regimens including ifosfamide in patients with newly diagnosed sarcomas. J Pediatr Hematol Oncol 22 (2): 112-8, 2000 Mar-Apr. [PUBMED Abstract]
Hartmann JT, Fels LM, Franzke A, et al.: Comparative study of the acute nephrotoxicity from standard dose cisplatin +/- ifosfamide and high-dose chemotherapy with carboplatin and ifosfamide. Anticancer Res 20 (5C): 3767-73, 2000 Sep-Oct. [PUBMED Abstract]
Skinner R, Cotterill SJ, Stevens MC: Risk factors for nephrotoxicity after ifosfamide treatment in children: a UKCCSG Late Effects Group study. United Kingdom Children's Cancer Study Group. Br J Cancer 82 (10): 1636-45, 2000. [PUBMED Abstract]
Stöhr W, Paulides M, Bielack S, et al.: Ifosfamide-induced nephrotoxicity in 593 sarcoma patients: a report from the Late Effects Surveillance System. Pediatr Blood Cancer 48 (4): 447-52, 2007. [PUBMED Abstract]
Oberlin O, Fawaz O, Rey A, et al.: Long-term evaluation of Ifosfamide-related nephrotoxicity in children. J Clin Oncol 27 (32): 5350-5, 2009. [PUBMED Abstract]
Daetwyler E, Bargetzi M, Otth M, et al.: Late effects of high-dose methotrexate treatment in childhood cancer survivors-a systematic review. BMC Cancer 22 (1): 267, 2022. [PUBMED Abstract]
Mitus A, Tefft M, Fellers FX: Long-term follow-up of renal functions of 108 children who underwent nephrectomy for malignant disease. Pediatrics 44 (6): 912-21, 1969. [PUBMED Abstract]
Bölling T, Ernst I, Pape H, et al.: Dose-volume analysis of radiation nephropathy in children: preliminary report of the risk consortium. Int J Radiat Oncol Biol Phys 80 (3): 840-4, 2011. [PUBMED Abstract]
Peschel RE, Chen M, Seashore J: The treatment of massive hepatomegaly in stage IV-S neuroblastoma. Int J Radiat Oncol Biol Phys 7 (4): 549-53, 1981. [PUBMED Abstract]
Ritchey ML, Green DM, Thomas PR, et al.: Renal failure in Wilms' tumor patients: a report from the National Wilms' Tumor Study Group. Med Pediatr Oncol 26 (2): 75-80, 1996. [PUBMED Abstract]
Paulino AC, Wilimas J, Marina N, et al.: Local control in synchronous bilateral Wilms tumor. Int J Radiat Oncol Biol Phys 36 (3): 541-8, 1996. [PUBMED Abstract]
Cheng JC, Schultheiss TE, Wong JY: Impact of drug therapy, radiation dose, and dose rate on renal toxicity following bone marrow transplantation. Int J Radiat Oncol Biol Phys 71 (5): 1436-43, 2008. [PUBMED Abstract]
Hoffmeister PA, Hingorani SR, Storer BE, et al.: Hypertension in long-term survivors of pediatric hematopoietic cell transplantation. Biol Blood Marrow Transplant 16 (4): 515-24, 2010. [PUBMED Abstract]
Abboud I, Porcher R, Robin M, et al.: Chronic kidney dysfunction in patients alive without relapse 2 years after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 15 (10): 1251-7, 2009. [PUBMED Abstract]
Ellis MJ, Parikh CR, Inrig JK, et al.: Chronic kidney disease after hematopoietic cell transplantation: a systematic review. Am J Transplant 8 (11): 2378-90, 2008. [PUBMED Abstract]
Poppe MM, Tai A, Li XA, et al.: Kidney Disease in Childhood Cancer Survivors Treated With Radiation Therapy: A PENTEC Comprehensive Review. Int J Radiat Oncol Biol Phys 119 (2): 560-574, 2024. [PUBMED Abstract]
Green DM, Wang M, Krasin MJ, et al.: Long-term renal function after treatment for unilateral, nonsyndromic Wilms tumor. A report from the St. Jude Lifetime Cohort Study. Pediatr Blood Cancer 67 (10): e28271, 2020. [PUBMED Abstract]
Green DM, Wang M, Krasin MJ, et al.: The accuracy of estimating equations for the evaluation of kidney function in adult survivors of unilateral, nonmetastatic, non-syndromic Wilms tumor: A pilot study from the St. Jude Lifetime Cohort Study. Pediatr Blood Cancer 72 (1): e31409, 2025. [PUBMED Abstract]
Breslow NE, Collins AJ, Ritchey ML, et al.: End stage renal disease in patients with Wilms tumor: results from the National Wilms Tumor Study Group and the United States Renal Data System. J Urol 174 (5): 1972-5, 2005. [PUBMED Abstract]
Hamilton TE, Ritchey ML, Haase GM, et al.: The management of synchronous bilateral Wilms tumor: a report from the National Wilms Tumor Study Group. Ann Surg 253 (5): 1004-10, 2011. [PUBMED Abstract]
Wu NL, Chen Y, Dieffenbach BV, et al.: Development and Validation of a Prediction Model for Kidney Failure in Long-Term Survivors of Childhood Cancer. J Clin Oncol 41 (12): 2258-2268, 2023. [PUBMED Abstract]
Hudson MM, Ness KK, Gurney JG, et al.: Clinical ascertainment of health outcomes among adults treated for childhood cancer. JAMA 309 (22): 2371-81, 2013. [PUBMED Abstract]
Landier W, Armenian SH, Lee J, et al.: Yield of screening for long-term complications using the children's oncology group long-term follow-up guidelines. J Clin Oncol 30 (35): 4401-8, 2012. [PUBMED Abstract]
Riachy E, Krauel L, Rich BS, et al.: Risk factors and predictors of severity score and complications of pediatric hemorrhagic cystitis. J Urol 191 (1): 186-92, 2014. [PUBMED Abstract]
Kersun LS, Wimmer RS, Hoot AC, et al.: Secondary malignant neoplasms of the bladder after cyclophosphamide treatment for childhood acute lymphocytic leukemia. Pediatr Blood Cancer 42 (3): 289-91, 2004. [PUBMED Abstract]
Frobisher C, Gurung PM, Leiper A, et al.: Risk of bladder tumours after childhood cancer: the British Childhood Cancer Survivor Study. BJU Int 106 (7): 1060-9, 2010. [PUBMED Abstract]
Ritchey M, Ferrer F, Shearer P, et al.: Late effects on the urinary bladder in patients treated for cancer in childhood: a report from the Children's Oncology Group. Pediatr Blood Cancer 52 (4): 439-46, 2009. [PUBMED Abstract]
Rosenbaum DH, Cain MP, Kaefer M, et al.: Ileal enterocystoplasty and B12 deficiency in pediatric patients. J Urol 179 (4): 1544-7; discussion 1547-8, 2008. [PUBMED Abstract]
Dietz AC, Seidel K, Leisenring WM, et al.: Solid organ transplantation after treatment for childhood cancer: a retrospective cohort analysis from the Childhood Cancer Survivor Study. Lancet Oncol 20 (10): 1420-1431, 2019. [PUBMED Abstract]
Actualizaciones más recientes a este resumen (05/02/2025)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha arriba indicada.
Se revisó el texto para indicar que los carcinomas queratinocíticos son algunas de las neoplasias subsiguientes más comunes entre los sobrevivientes de cáncer infantil y exhiben una fuerte relación con la radioterapia (se citó a Boull et al. como referencia 83).
Se revisó el texto para indicar que en un análisis actualizado del Childhood Cancer Survivor Study (CCSS) se confirmó un aumento del riesgo de carcinomas queratinocíticos en aquellos participantes que en el pasado habían recibido radioterapia. En comparación con los participantes que no recibieron radioterapia, los participantes del CCSS tratados con radioterapia presentaron un riesgo 4,5 veces mayor de carcinomas queratinocíticos. También se añadió texto para indicar que el riesgo de carcinomas queratinocíticos aumentó a medida que aumentaron las dosis de radioterapia. Cada 10 Gy de radioterapia en el sitio del carcinoma queratinocítico, el riesgo relativo (RR) fue similar para los sitios en cabeza y cuello, y extremidades. El RR fue levemente superior en los sitios del tronco. Los trasplantes de células madre hematopoyéticas (TCMH) alogénicos y autógenos se relacionaron con un aumento del riesgo de carcinomas queratinocíticos
Se revisó el texto para indicar que los factores de riesgo del melanoma maligno incluyen la radioterapia de más de 40 Gy, los alquilantes y la bleomicina (se citó a Rotz et al. como referencia 85).
Se añadió texto sobre los resultados de un estudio en el que se evaluó la incidencia, los factores de riesgo y los desenlaces de pacientes con melanoma en la cohorte del CCSS.
Se añadió texto sobre los resultados de un estudio Dutch Childhood Cancer Survivors Study LATER 2 en el que se evaluó la prevalencia y los determinantes del síndrome metabólico en una cohorte de 2338 sobrevivientes adultos de cáncer infantil, en comparación con 132 226 adultos sin antecedentes de cáncer de la cohorte de Lifelines (se citó a Bolier et al. como referencia 102).
Se añadió texto sobre los resultados de un estudio en el que se evaluó el déficit de densidad mineral ósea en 3919 sobrevivientes a 5 años en la cohorte del St. Jude Life (SJLIFE) (se citó a Goodenough et al. como referencia 35).
Se revisó el texto para indicar que el factor de tratamiento más importante que confiere riesgo de osteonecrosis es la exposición prolongada a los corticoesteroides, que es característica en los regímenes usados para la leucemia linfocítica aguda (LLA), el linfoma de Hodgkin, el linfoma no Hodgkin y el TCMH (se citó a Giertz et al. como referencia 56).
Se añadió texto para indicar que los adultos sobrevivientes de un tumor de Wilms unilateral, no metastásico y no sindrómico, tratados con radioterapia dirigida a todo el abdomen, presentan riesgo de deterioro del funcionamiento renal. También se añadió texto para indicar que en un estudio piloto de la cohorte del SJLIFE se evaluó la precisión de las ecuaciones que evalúan el funcionamiento renal en este grupo de pacientes (se citó a Green et al. como referencia 35).
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre los efectos tardíos del tratamiento anticanceroso en la niñez. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El consejo editorial del PDQ sobre el tratamiento pediátrico, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
Si el artículo se debe analizar en una reunión del consejo.
Si conviene añadir texto acerca del artículo.
Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Efectos tardíos del tratamiento anticanceroso en la niñez son:
Sally J. Cohen-Cutler, MD, MS (Children's Hospital of Philadelphia)
Louis S. Constine, MD (James P. Wilmot Cancer Center at University of Rochester Medical Center)
Nita Louise Seibel, MD (National Cancer Institute)
Emily S. Tonorezos, MD, MPH (National Cancer Institute)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El consejo editorial del PDQ sobre el tratamiento pediátrico emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como “En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]”.
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
PDQ® sobre el tratamiento pediátrico. PDQ Efectos tardíos del tratamiento anticanceroso en la niñez. Bethesda, MD: National Cancer Institute. Actualización: <MM/DD/YYYY>. Disponible en: https://www.cancer.gov/espanol/tipos/infantil/efectos-tardios-pro-pdq. Fecha de acceso: <MM/DD/YYYY>.
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia, las opciones de tratamiento se clasifican como “estándar” o “en evaluación clínica”. Estas clasificaciones no se deben utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.



 según el tipo de segundo cáncer: sarcoma, cáncer de piel (CCB), meningioma, cáncer de glándulas salivales, glioma, cáncer de mama y cáncer de tiroides.")