Inhibition of the MAPK pathway induces re-localization of membranous proteins, leading to adaptive resistance to KRAS G12C inhibitor mediated by YAP-induced MRAS
, by Yuta Adachi and Hiromichi Ebi

Yuta Adachi is a staff scientist in the laboratory of Hiromichi Ebi at the Aichi Cancer Center Research Institute in Nagoya, Japan. His research is focused on interrogating mechanisms by which KRAS mutant tumor cells escape from mutant specific KRAS inhibitors.
Hiromichi Ebi is Chief of the Division of Molecular Therapeutics at the Aichi Cancer Center Research Institute and Director of the Precision Medicine Center at Aichi Cancer Center Hospital. He is also Adjunct Professor of the Division of Advanced Cancer Therapeutics at the Nagoya University Graduate School of Medicine. In his post-doctoral work, Dr. Ebi studied strategies to improve the efficacy of molecular targeting agents in the laboratory of Dr. Jeffrey Engelman at the Massachusetts General Hospital Cancer Center.
The recent development of KRAS G12C inhibitors has led to a paradigm shift in the treatment of these RAS mutant cancers. However, the rapid development of drug resistance suggests that this is likely to be a recurring theme in targeted therapy. Therefore, determining the mechanisms by which KRAS mutant tumor cells escape inhibitory insults, and devising rationale combination strategies to combat innate and acquired resistance will be critical towards improving tumor response and patient outcomes.
Resistance to cancer therapy mainly consists of primary/intrinsic and acquired. While primary/intrinsic resistance is caused by the presence of drug resistant clones, acquired resistance arises from the selective expansion of pre-existing, rare, fully resistant subpopulations, or from the emergence of drug resistant clones incited by therapeutic pressure. These resistant clones can develop from genetic mechanisms, such as mutations and amplifications, or from non-genetic mechanisms, such as drug-induced rewiring of gene-expression, signaling, and metabolic networks that decrease a tumor cell’s dependence on the targeted oncoprotein. In the case of acquired resistance to KRAS G12C inhibitors, secondary mutations in RAS and genomic alterations in the MAPK signaling cascade have been detected 1,2. We previously demonstrated that epithelial-to-mesenchymal transition (EMT) is a cause of both intrinsic and acquired resistance to KRAS G12C inhibitors 3.
Adaptive resistance is in the middle of intrinsic and acquired, wherein dampening of the therapeutic response is precipitated by changes in the signaling network following treatment, occurring within hours of drug exposure 4. Adaptive resistance is a major contributor to the insensitivity of tumor cells to targeted therapies that block the MAPK pathway, as this network is extensively regulated by homeostatic negative feedback mechanisms that fine-tune signaling output in both cancer and normal cells.
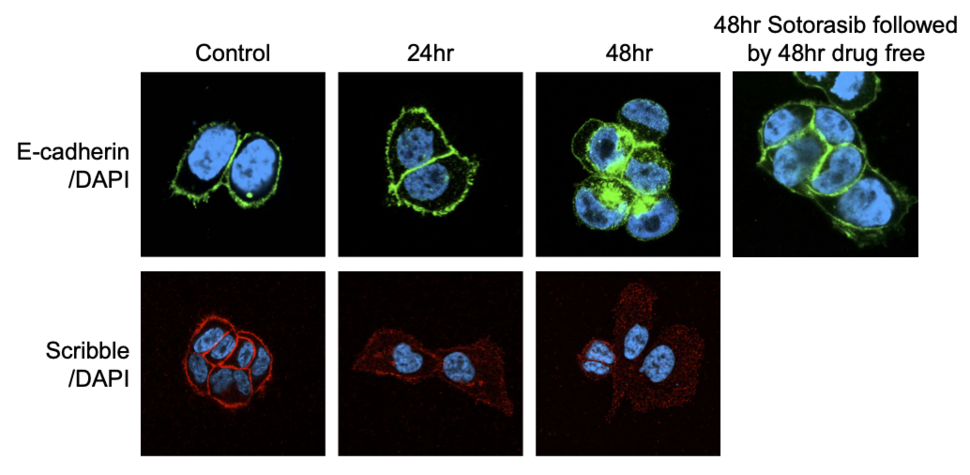
Figure 1. The KRAS G12C inhibitor sotorasib induces re-localization of membranous proteins E-cadherin and Scribble.
In our recent study, we initially observed that the EMT process begins shortly after treatment with KRAS G12C inhibitors – within 48 hours 5. KRAS G12C inhibition leads to the re-localization of E-cadherin and the apical-polarity protein Scribble from the plasma membrane to the cytosol (Figure 1). The induction of EMT was not caused by transcriptional regulation of EMT transcription factors, but rather by the suppression of MAPK signaling. Our preliminary analysis suggests that MAPK signaling upregulates protein levels of ZDHHC7, which promotes the palmitoylation of Scribble, required for its protein trafficking and membrane localization. Notably, the process was reversed by sotorasib withdrawal.
Scribble positively regulates the Hippo pathway kinases, and its downregulation leads to YAP/TAZ activation in mammary cells 6. Treatment with KRAS G12C inhibitors also downregulated Hippo pathway signaling, leading to the nuclear translocation of YAP proteins and their transcriptional activity. The combination of sotorasib with a TEAD inhibitor achieved tumor shrinkage in vivo, and this data is supported by an accompanying paper 7.
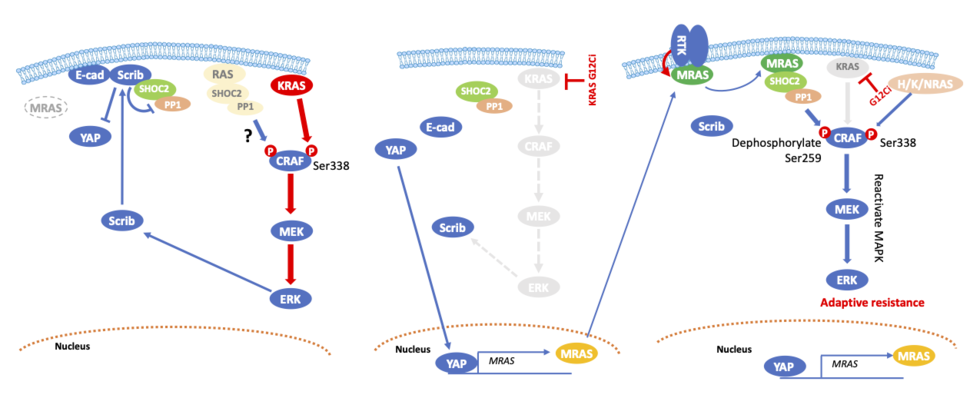
Figure 2. Schematic diagram of reactivation of MAPK signaling following KRAS inhibition. left. In the absence of drug, mutant KRAS activates MAPK signaling, which promotes the membrane localization of Scribble (Scrib). Scribble forms a complex with SHOC2/PP1c and inhibits the nuclear translocation of YAP. Middle. KRAS G12C inhibition downregulates MAPK activation, which leads to a reduction in the membranous localization of Scribble. Since Scribble cannot inhibit YAP, YAP translocates to the nucleus and transcribes its downstream proteins including MRAS. Right. MRAS translocates to the membrane where it is activated by RTKs. GTP-MRAS forms a complex with SHOC2/PP1a (the MSP complex) that dephosphorylates CRAF S259. Following CRAF dephosphorylation, it is likely that H/K/NRAS phosphorylate CRAF S338, resulting in reactivation of MAPK signaling.
Furthermore, we discovered that MRAS is a direct target of YAP. Although MRAS is a RAS superfamily gene that is structurally similar to H/K/NRAS, and shares an almost identical effector binding sequence at switch I 8,9, MRAS mediates its signaling through SHOC2, a positive regulator of the RAS pathway with by far the strongest binding affinity for it among members of the RAS family 10. Activated MRAS binds to SHOC2 and the catalytic subunit of PP1, recruiting them to the plasma membrane where the ternary complex dephosphorylates CRAF at S259, resulting in CRAF activation. Since MRAS expression is low at the basal level, Scribble interacts with SHOC2 to inhibit SHOC2-mediated dephosphorylation of CRAF S259. KRAS inhibition induces a molecular switch from Scribble/SHOC2 to MRAS/SHOC2, leading to reactivation of MAPK signaling by dephosphorylating CRAF S259. Given that feedback reactivation of MAPK signaling following suppression of KRAS G12C is mediated by receptor tyrosine kinases (RTKs), inhibitors against RTKs or adaptor proteins such as SOS1 and SHP2 downregulated MRAS-GTP (Figure 2).
Based on our findings, we propose that protein re-localization is a novel, non-genetic mechanism of resistance. While EMT has long since been demonstrated as a key mechanism of resistance, it is surprising how quickly the reorganization of membranous proteins is induced following treatment with KRAS G12C inhibitors. Additionally, the re-localization of these membranous proteins is reversible, which is consistent with current notions that EMT is not a binary state, but rather a complicated and dynamic transitional state between the epithelial and mesenchymal phenotypes 11,12. Since MAPK signaling is involved in the regulation of Scribble localization, re-localization of this factor is extensively observed following treatment with molecular targeting agents in oncogene addicted cancers with aberrantly activated MAPK signaling. Further investigations should be made to determine if additional mechanisms can induce protein re-localization following inhibition of mutant KRAS.
YAP is known to induce mutant KRAS independency 13,14, and it’s also involved in the resistance mechanism of oncogene addicted cancers to targeted therapies 15-18. In the case of KRAS G12C mutant cancers, activated YAP plays a key role in the intrinsic, adaptive, and acquired resistance mechanism of these tumor cells to KRAS G12C inhibitors that is associated with the mesenchymal phenotype. It would be interesting to see if the combination of a KRAS G12C inhibitor with a TEAD inhibitor will be tolerable and effective in the clinic. Notably, while MRAS knockdown prevents adaptive resistance to KRAS G12C inhibitors, knockdown of MRAS has little effect on the efficacy of these inhibitors once full EMT is established. This suggests that YAP rewrites many other signaling networks, including apoptotic and metabolic processes.
Our findings provide functional details to a recent series of studies that determined the structure of the MSP (MRAS-SHOC2-PP1C) complex 19-22. The active MSP complex formed only in the presence of GTP-bound MRAS, but its activity was downregulated by inhibition of RTKs or adapter proteins, suggesting that MRAS is the hub of feedback activation of MAPK signaling via RTKs. In line with our findings, deletion of SHOC2 sensitized KRAS mutant cells to MEK inhibitors and EGFR mutant cells to EGFR tyrosine kinase inhibitors in NSCLC 23-25.
The relative druggable nature of the MSP complex warrants therapeutic development targeting this complex to prevent feedback reactivation of MAPK signaling. However, the precise role of this complex, and its relationship with other RAS isoforms, must first be established. In the basal state, to prevent inadvertent activation of the MAPK pathway, CRAF is phosphorylated at residues S259 and S621, which promotes the binding of dimeric 14-3-3 proteins that sequester RAF, restricting it to the cytoplasm. Once MRAS is activated, RAS-RAF binding is thought to displace 14-3-3 proteins from the CRAF S259 site, permitting its dephosphorylation by PP1C in the MSP complex. Dephosphorylation of CRAF S259 by the MSP complex precedes catalytic activation of S338, and this appears to be the first step in activating the MAPK pathway. However, epithelial marker positive KRAS mutant cancer cells express very low levels of MRAS. Even though the lower affinity SHOC2-H/K/NRAS-PP1C complexes are able to function in some situations 26, the mechanism by which CRAF S259 is dephosphorylated in MRAS-low expressing cells is unclear. In the NCI-H358 KRAS G12C mutant lung cancer cell line, MRAS knockout upregulated ERK phosphorylation, presumably due to compensation by other RAS isoforms 5.
Tumor cells use every possible means to evade targeted drugs. Our study highlights the importance of post-translational regulation, such as protein localization, as a new mechanisms of resistance to molecular targeted drugs. YAP is a potential kryptonite for KRAS mutant cancer cells, although targeting this factor might prove toxic in patients. As a key mediator of feedback reactivation of MAPK signaling, the MSP complex may be an attractive drug target to prevent adaptive resistance to KRAS G12C inhibitors.