Engaging patients, advocates, and researchers to improve the lives of young people with rare cancers
The MyPART (My Pediatric and Adult Rare Tumor) Network
-
 The MyPART Network
The MyPART NetworkLearn more about the MyPART group of scientists, patients, family members, advocates, and health care providers working on rare tumors.
-
 Participate in Our Trial
Participate in Our TrialHelp speed up the development of new treatments by giving researchers the tools they need.
-
 Why We Study Rare Cancers
Why We Study Rare CancersRare cancers make up about a quarter of all cancer cases. Learn why we are committed to studying these cancers.

NCI Rare Tumor Clinics
The MyPART team hosts clinics at the NIH Clinical Center where patients and experts meet to learn more about the tumor from each other.
Learn MoreResearch on Rare Tumors
-
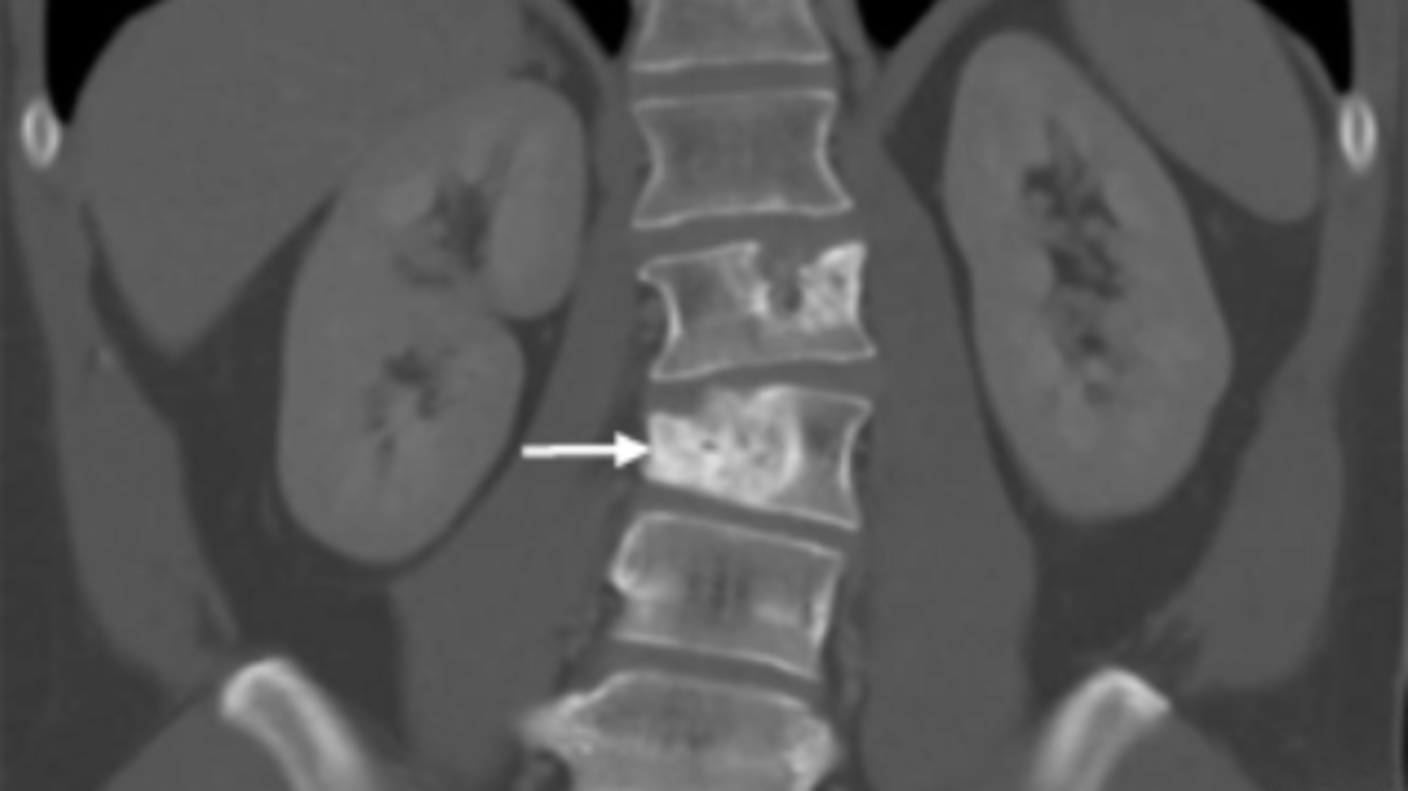 Study Confirms Atezolizumab’s Effectiveness Against a Rare Sarcoma
Study Confirms Atezolizumab’s Effectiveness Against a Rare SarcomaFor people with alveolar soft part sarcoma, immunotherapy drug offers new option.
-
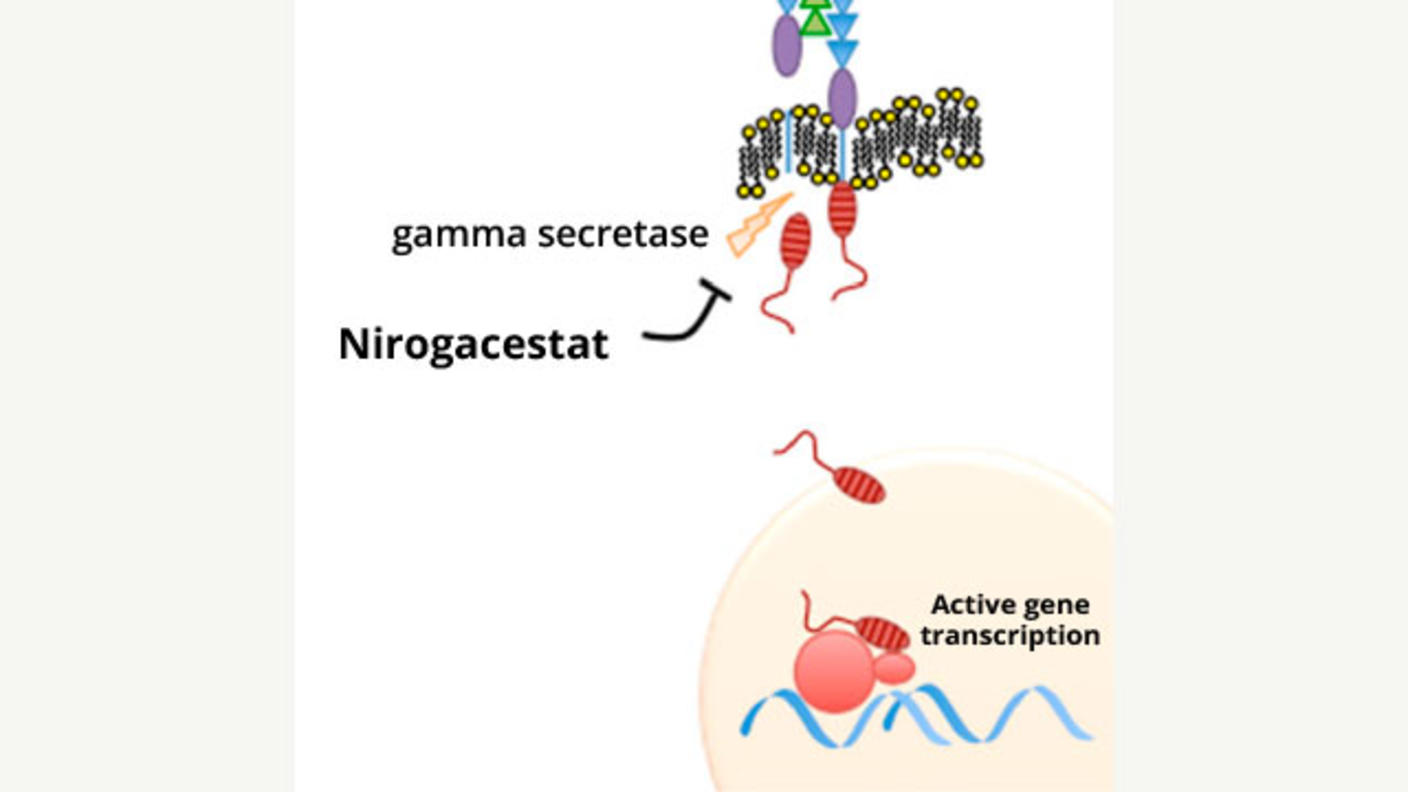 A Potentially Major Advance for Treating Desmoid Tumors
A Potentially Major Advance for Treating Desmoid TumorsNirogacestat treatment shrinks tumors, improves progression-free survival, and reduces pain.
-
 Selumetinib Approved by FDA to Treat Children with NF1
Selumetinib Approved by FDA to Treat Children with NF1NCI-led trial showed drug shrank tumors, improved quality of life.