Meticulous Reconstructions of Tumor Origins With Single-Cell DNA Sequencing: Researcher Interview with Nicholas Navin
, by Peggy I. Wang
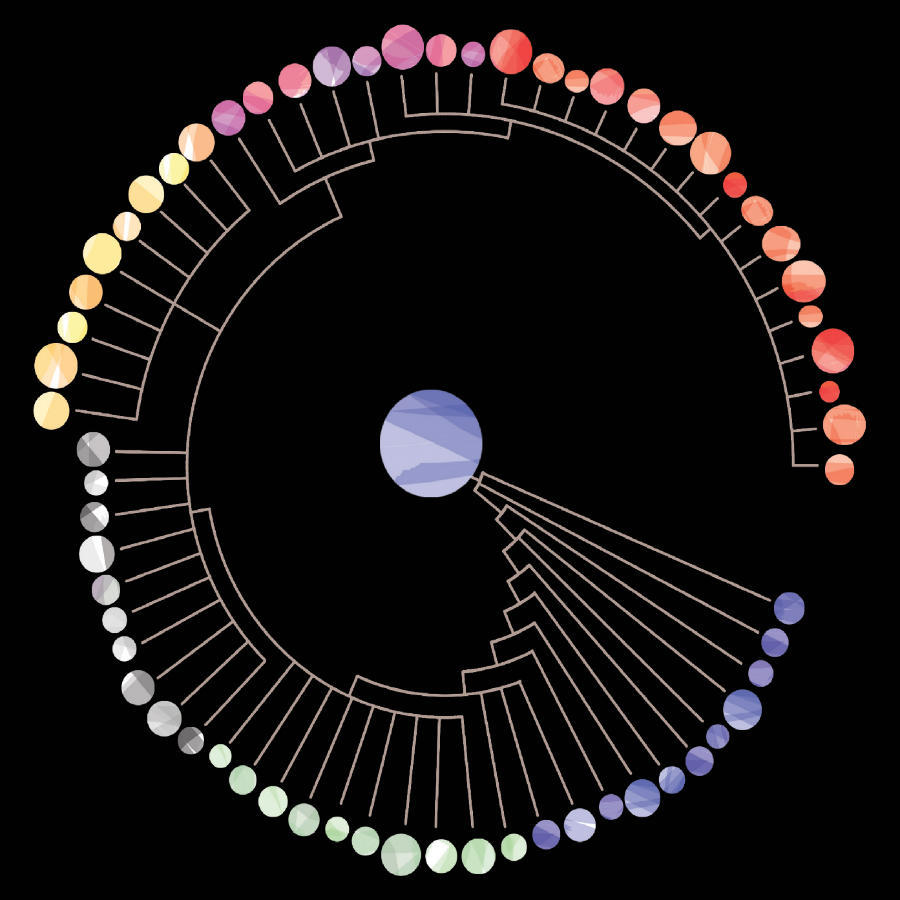
A phylogenetic lineage in which single cells (circles) from a primary tumor evolved for a period of time before diverging to form a metastatic lineage. The colors indicate major clones with different genotypes.
Credit: Aislyn Schalck and Nicholas Navin
Dr. Nicholas Navin pioneered single-cell DNA sequencing—and did so as a side project in his Ph.D. work. Now an associate professor at The University of Texas MD Anderson Cancer Center, he continues to develop single-cell methods to study cancer evolution during invasion, metastasis, and therapy resistance.
I spoke with Dr. Navin about the technology and his research, considerations for working in the single-cell sequencing space, and what’s next for his lab.
Peggy I. Wang: What originally motivated you to work on single-cell DNA sequencing (scDNA-seq)?
Nicholas Navin: I was a PhD student at the time, flow sorting cells and using copy number microarrays to look at copy number evolution. I did hundreds of tumors that way. I was very frustrated because no matter how much I macrodissected a tumor or flow sort out populations, it was always a mixture of cells. I never knew, when I was looking at genomic information, is this one cell or a mix of 20 to 30 different genotypes. I think out of that frustration, I worked harder and harder to get single-cell methods to work. That was the ultimate resolution that could give you a clear answer about how genomes evolve over time.
PIW: Could you give a quick rundown on how scDNA-seq works?
NN: It involves three steps: (1) Isolating the cell (which can be done using high-throughput methods), (2) genome amplification followed by sequencing library construction, and (3) sequencing the single cell libraries, which is mainly performed using Illumina sequencers due to their high data output and low cost-per-case to maximize coverage depths in single cells.
As an overall strategy, the cost of a single-cell genome is the cost of a whole genome, so it’s not economically feasible to profile lots of cells that way. So we’ve moved from that towards more targeted methods, looking at cancer gene panels, but then we realized that’s quite wasteful, too. Really what you want to do is know all the mutations of interest beforehand and create a custom panel and sequence exactly the sites you’re interested in and look at thousands or tens of thousands of cells.
PIW: Are there different flavors of the technology? How might they be used differently?
NN: We use several different platforms for isolating the cells: FACS, micromanipulation, microfluidics, and nanowells. Using microfluidics is cheaper and more high-throughput, micromanipulation allows you to pick out a rare cell, and FACS is somewhere in between with 384 or 1536 well plates. With nanowells, you can image the cells and pick which ones you want to analyze.
As for whole-genome amplification, different chemistries such as DOP-PCR (good for copy number estimation), multiple-displacement-amplification (higher coverage for mutation detection) or tagmentation (useful for higher-throughput copy number profiling) are available.
We also use a spatially-resolved method to see where the cells are located in tissue sections before we use laser capture microdissection to cut out single cells. With this method, we can see things like where cells are in breast tissue ducts and how far away they have migrated.
PIW: Is scDNA-seq accessible to a typical research lab?
NN: It’s becoming more and more accessible, with commercial products becoming available just as of a few months ago. ScDNA-seq has been more in the hands of expert labs for quite a while, but just recently now it’s becoming more commercialized with copy number profiling and mutations. I’m quite interested to see what happens as people start to use these methods to study cancer and also beyond cancer, such as genetic diversity in the brain. I think it’s a pretty exciting time right now.
PIW: What are some considerations or challenges for analyzing scDNA-seq?
This topic warrants a whole other discussion, but one major consideration is allelic dropout, which leads to lots of false negatives. Some of these errors can be overcome by grouping data together, since most errors are random, using either clustering or phylogenetic approaches which is where the thinking is going now. Machine learning approaches are also being used more and more to filter out cells with lots of errors or classify cell types or clones.
PIW: What aspects about cancer can you study with single-cell sequencing that you could not with bulk?
NN: One goal we have is to understand premalignant breast cancers: How does a single cell in a duct actually give rise to a premalignant lesion? And how do the cells, once they expand in the duct, cross the basement membrane and establish the invasive cancer? What’s really important here are spatially-resolved methods, which is one area we’ve been focusing on for technology development.
Another area we’re studying is metastasis: does a single cell really seed a metastatic tumor, or does it take multiple cells? When does dissemination happen; is it early on in the lifetime of the primary tumor or is it a relatively late event? What are the migration trajectories of cells as they seed across multiple organ sites? We study this both by looking tumor circulating cells in the blood and also looking at matched primary and metastatic tumors.
A third area is therapeutic resistance: why do certain patients respond to therapy and others do not? Can that be predicted based on the tumor cells or microenvironment? And for that, we’re trying to leverage longitudinal samples, like liquid biopsies, where it’s feasible to get 10-20 samples longitudinally from a patient, and also looking at matched fine needle aspiration and core biopsies and surgical samples.
PIW: How might single-cell technologies in general change things for patients?
NN: Our work could inform treatment strategies and improve diagnostics tools for any kind of cancer. Breast cancer is our main focus, especially triple negative breast cancers, because they really have limited therapeutic options and genetically, they have diverse and complex aneuploid genomes. So there’s a big need to develop better diagnostics and therapeutic options.
Do we need to target the truncal mutations? Those are the early, initiating mutations in the evolutionary tree—but those mutations might not be as important later on as the tumor is expanding. So more of these evolutionary trees are being used to think about how to target the most malignant population or the one most likely to become metastatic and not necessarily target all the cells.
Also, what’s exciting about liquid biopsy is that it really is feasible to track not just single mutations, but all the mutations and all the copy number alterations in patients over time, after every cycle of therapy, and also when they switch therapies. We can start to understand what’s happening at the genomic level when a resistance emerges.

Dr. Nicholas Navin, Associate Professor, Department of Genetics at University of Texas MD Anderson Cancer Center
PIW: What are some of the big questions you’re most excited to answer in your work now?
NN: Is aneuploidy a cause or consequence of tumor progression? This question has been out there a long time. So I’m very interested in copy number evolution and trying to figure out when it occurs during the life-cycle of a breast tumor.
Clonal diversity and intratumoral heterogeneity: is that just noise, or is it actually very important for the selection and evolution of tumors?
Does a tumor really evolve from one single cell, or are there also cases where multiple cells can initiate and give rise to a tumor? For multi-focal tumors, are there multiple cells that go on to form a tumor or just one cell?
Another area that I think is pretty interesting is trying to combine imaging technologies with single-cell genomics. For a long time, my lab has been going from genotype to phenotype and I’m interested in going the other way around: using live-cell imaging or another imaging method to observe morphologies and more phenotypic characteristics and going back to single-cell genomes and transcriptomes and trying to understand which pathways explain those features. Bringing these two fields together will be very powerful for understanding cancer.