Beat AML 1.0: A Collaborative Program for Functional Genomic Data Integration
, by Jeffrey W. Tyner, Ph.D.
By Subhashini Jagu, Ph.D.
Scientific Program Manager for the Cancer Target Discovery and Development (CTD2) Network
The largest-to-date dataset on primary acute myeloid leukemia (AML) samples offering genomic, clinical, and drug response data was recently published in Nature. This valuable resource for the research and patient communities was developed by Beat AML 1.0, a major collaborative effort led by Oregon Health and Science University (OHSU).
Beat AML 1.0 is an example of the possible productivity of team science; the dedicated work of over 100 researchers from 11 centers over five years contributed to the program’s success. Dr. Jeffrey Tyner, associate professor of cell, developmental and cancer biology in the OHSU School of Medicine, describes the program in detail.
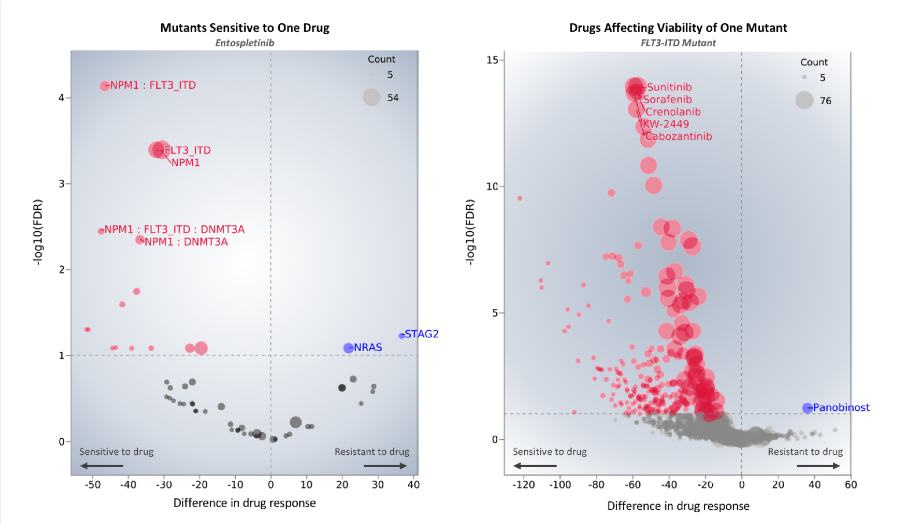
Visualizations of drug sensitivity data highlighting a known interaction between a FLT3 mutation and the inhibitor Entospletinib. Further visualizations of Beat AML drug sensitivity screening, genomic, and clinical data are available in Vizome.
Program Rationale: Four Decades Without Progress for a Common Cancer
Acute myeloid leukemia (AML) is one of the most common forms of hematologic malignancies with a standard chemotherapy regimen that has not seen significant change for ~40 years. Challenges associated with drug development for AML have included complex mutational patterns within and across patients as well as a lack of understanding of pharmacologic agents that can effectively be employed for most mutational events. To better understand drugs and drug combinations that should be prioritized for further development within clinical and/or molecular subsets of AML patients, we developed the Beat AML 1.0 research program.
Program Overview: An Extensive Genomic, Clinical, and Drug Dataset
The Beat AML program involved 11 academic medical centers who worked collectively to accrue a cohort of ~950 AML patient specimens and 11 pharmaceutical and biotechnology companies, who supplied drugs for testing. These specimens were subjected to whole exome sequencing, RNA-sequencing, and ex vivo drug sensitivity analyses. The majority of this dataset was published in October 2018 in Nature.
This manuscript reports on 672 patient specimens, including 622 specimens with exome data, 451 with RNA-sequencing, and 409 specimens where drug sensitivity data can be coupled with exome and/or RNA-sequencing. In addition, detailed clinical annotations were collected representing over 30,000 data points regarding demographic and diagnostic information, hematology/chemistry labs, extracted fields from pathology and cytogenetics reports, and treatments/outcomes.
Accessing the Data: Multiple Options for Visualization and Analysis
The data from this cohort are available in several formats. All sequencing data are being submitted to NCI's Genomic Data Commons and drug sensitivity data will be available in the CTD2 Data Portal. Processed summaries of all data are found in the supplementary information file associated with the manuscript. A data exploration and visualization tool, Vizome, has been constructed around this project and is publicly available offering a point-and-click interface for mining the dataset.
Summary of Findings: Insights into Patient-Specific Drug Sensitivity
In our manuscript, we report a series of strategies for integration of these data to inform markers of drug sensitivity or resistance. We examined all recurrent mutations (and some combinations of mutations) to determine statistically significant differential drug sensitivity compared with cases that were wild-type for the mutation(s) in question.
These analyses identified many associations between mutational events and drugs worth further investigation. For example, cases with TP53 or ASXL1 mutations were resistant to a broad panel of drugs but sensitive to elesclomol and panobinostat, respectively. Cases with FLT3 internal tandem duplications demonstrated sensitivity to entospletinib, a previously suggested interaction.
We also examined the 20% most and least sensitive subset of samples to each drug that were then used for differential expression analyses, with approximately two-thirds of the drugs tested showing a gene expression signature that could distinguish sensitive from resistant samples in a significant manner.
Drug sensitivity was also correlated with a variety of clinical parameters, such as de novo AML versus AML that transformed from a setting of myelodysplasia, and differential drug sensitivity was observed for numerous drugs in these comparisons
A weighted gene correlation network analysis was also performed to identify clusters of genes with similar patterns of expression. These clusters as well as recurrent mutations were analyzed against drug sensitivity as a continuous variable to understand correlations between mutations, gene expression, and drug sensitivity simultaneously.

Dr. Jeffrey W. Tyner, associate professor of Cell, Developmental and Cancer Biology at Oregon Health and Science University.
Collectively, these analyses offered both proof-of-principle findings that confirmed the validity of the analytical strategy as well as many novel correlations and potential biomarkers for drug sensitivity.
Next Steps: Novel Analyses and Expansion of the Data Planned for Beat AML 2.0
There is much ongoing work to build on this program. Some of this work aims to leverage the dataset in new ways and includes additional analytics of the data (e.g., copy number, clonality, splicing, germline mutations, pathway enrichment, etc.), new ways of integrating the data to make additional predictions for markers of drug sensitivity or resistance and understanding the contribution of the microenvironment to drug response patterns.
Efforts are also underway to expand the dataset, including collecting methylation, proteomics, metabolomics, and secreted factors data, as well as characterizing new patient samples.
Work is also underway to explore mechanistic underpinnings of drug sensitivity and resistance biomarkers as well as further pre-clinical and clinical development of the most promising drug response signals.