Systems Cancer Immunology for the Masses
, by Erin F. Simonds, Ph.D., Edbert D. Lu, Ph.D., and William A. Weiss, Ph.D.
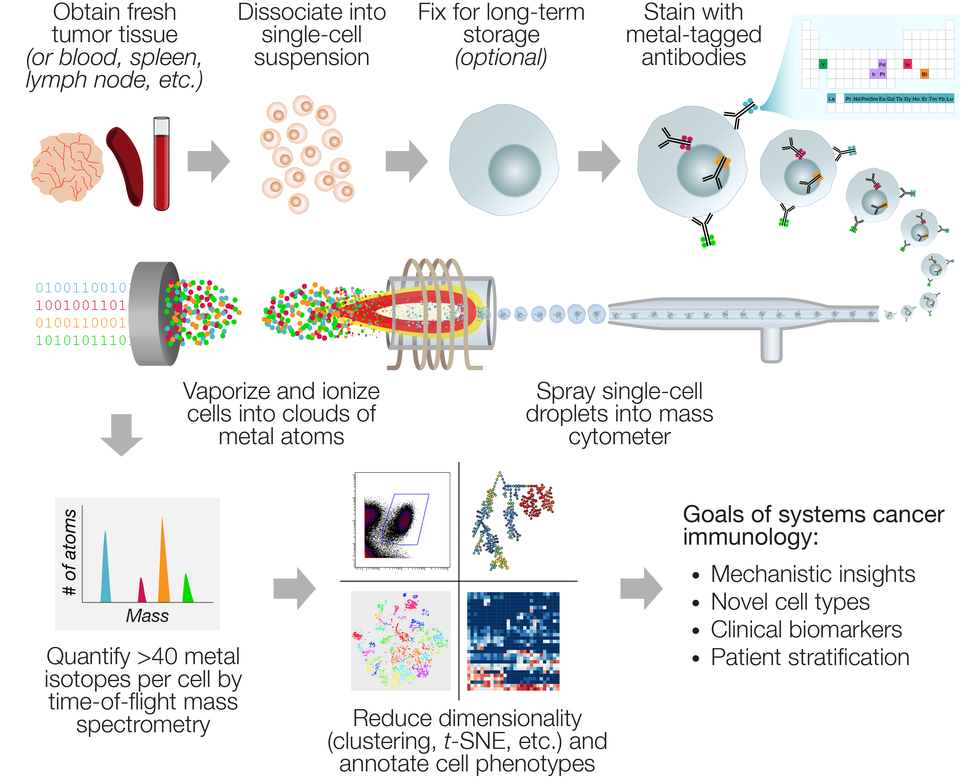
Mass cytometry analysis of tumor and immune tissues informs cancer systems immunology. The sample processing and antibody staining steps for mass cytometry are analogous to flow cytometry, except that metal-conjugated antibodies are used (top row). Inside the instrument, antibody-labeled cells are sprayed through a glass nebulizer and carried by a stream of argon into an inductively-coupled plasma (ICP) torch, where the metal atoms are liberated from the antibodies and directed as single-cell ion clouds into a mass spectrometer (middle row). The raw data from the mass spectrometer is converted into single-cell measurements of antibody abundance, which is then fed into a variety of computational tools to reveal cellular phenotypes and changes in marker expression (bottom row).
Credit: Erin F. Simonds
Cytometry by time-of-flight (CyTOF®), or generically, mass cytometry, is an antibody-based method of measuring cellular phenotypes in single-cell suspensions. This technology is unique in its ability to multiplex up to 44 distinct antibody-based markers, in millions of cells per day, at a low cost per cell, with minimal signal overlap between markers1. Mass cytometry achieves these improvements by using atomic mass spectrometry to measure metal-tagged antibodies, rather than lasers and fluorochrome-tagged antibodies, as in classical flow cytometry.
While mass cytometry is one of several available single-cell analysis platforms, it is particularly well-suited for immuno-oncology research. In immuno-oncology, researchers are often interested in the abundance and co-expression patterns of 30-50 markers of immune cell populations. Critically, these markers tend to be those for which flow cytometry-compatible antibodies have already been developed, therefore making them easily adaptable for mass cytometry. As mass cytometry can measure up to 44 antibody-based markers per cell, it opens up “systems cancer immunology” to the masses by formalizing and simplifying the analysis of diverse immune cell subsets across immune organs, tumor types, and drug treatments. For example, several groups have used mass cytometry to monitor T lymphocyte, B lymphocyte, natural killer, myeloid, and dendritic cell phenotypes simultaneously with 30-marker panels, leaving some bandwidth for novel or investigational markers. Consolidating all of these antibodies to a single staining cocktail simplifies sample processing and analysis across experimental conditions.
CyTOF® mass cytometry was developed in the mid-2000s by engineers at DVS Sciences in Toronto, Canada. In the years following, it has matured as a discipline with a wide user base. A third-generation instrument, the HeliosTM mass cytometer, was released by Fluidigm Corporation in 2015, a year after their acquisition of DVS Sciences. The first publications with this technology were in 2010 - 2011, in which mass cytometry was applied to studies of normal human immunology and hematopoiesis. Since 2015, numerous high-profile publications have featured mass cytometry analysis of a wide range of leukemias and cancer types, including mouse models of skin, colorectal, and breast cancer, as well as cohorts of human patients with AML, pre-B ALL, multiple myeloma, non-small cell lung cancer, clear cell renal carcinoma, melanoma, glioblastoma, and ovarian cancer.
Mass cytometry produces data with a unique combination of high-dimensionality and high-throughput compared to other technologies for single-cell profiling. The workflow (Figure) is similar to flow cytometry: Samples must first be processed into single-cell suspensions (i.e., enzymatic digestion of solid tumors), which can then be either measured immediately, cryopreserved as viable cells, or fixed and frozen. Cells are then stained with metal-conjugated antibodies against target markers, as well as metal-based dyes for viability, DNA content, and sample identification (“barcoding”). The specialized processing needed for flow or mass cytometry analysis of solid tumors requires more hands-on time and expertise than banking formalin-fixed or frozen tissue blocks, which has limited the use of this technology for existing pathology sample archives. However, as the field of immuno-oncology grows, prospective banking of single-cell suspensions is becoming more standardized and commonplace, particularly in the setting of mouse studies and clinical trials.
One challenge in mass cytometry experimental design is that the decision of ~40 metal-tagged antibodies must be made a priori. Metal-conjugated antibodies to commonly used targets in immunology are available commercially; others must be learned from publications or conjugated in-house. Because mass cytometry is an antibody-based approach, the range of potential analytes is largely limited by the availability of commercial antibody clones. Like flow cytometry, fluorescent microscopy, or qPCR, this inherently creates a “lamppost” situation in which the scope of discovery is limited to the set of preselected targets. Other single-cell approaches that cast a wider net, such as single-cell RNA-sequencing or DNA-tagged antibodies, fill the need for a broader look at an experimental system. However, these less-biased methods tend to be significantly more expensive on a per-cell basis. For example, scRNA-seq using the 10X Chromium™ system costs approximately $0.10 per cell.
In our hands, when using commercially available metal-conjugated antibodies and paying for use of a shared HeliosTM mass cytometer, a 40-parameter analysis of 100,000 cells costs about $300, or about $0.003 per cell. It is possible to further reduce costs by using metal-conjugated antibodies that are prepared in-house, although this requires more optimization. Access to mass cytometry is continually improving for institutions without the necessary instrumentation or expertise. Academic cores offering full-service mass cytometry sample preparation and analysis for outside users can now be found on four continents (a community-maintained list of cores can be found at cytoforum.stanford.edu). Building on the innovative detection modality behind mass cytometry, there are now other approaches available that use metal-tagged antibodies. High-parameter imaging-based platforms such as the IONPath MIBIScope™ I and Fluidigm Hyperion™, offer the opportunity to use archived tissue blocks. These emerging platforms maintain tissue morphology and include information about the spatial distribution of cell subsets (i.e. in blood vessels, lymph nodes, areas of necrosis), which is lost in single-cell suspensions of solid tissue.

Erin F. Simonds, Ph.D., Edbert D. Lu, Ph.D., and William A. Weiss, Ph.D. of UCSF Departments of Neurology, Neurological Surgery, and Pediatrics
The most challenging hurdle in the field of mass cytometry is the process of data analysis. Fortunately, in the last decade, the workflow for mass cytometry data analysis has become more accessible and streamlined. Moreover, the unique needs of the field have inspired new approaches to analyze this type of data.
Mass cytometry data can be thought of as a long table with a few dozen columns and millions of rows. A typical experiment may contain a table of this size for many patients or experimental conditions. In most cases, this scale of data exceeds conventional approaches for flow cytometry data analysis, such as dot plots or histograms. The breadth of up to 44 markers per cell also creates an opportunity to discover unanticipated combinations or expression patterns, so an unbiased analysis approach is often preferred.
A popular strategy has been to reduce the dimensionality of the data by collapsing multiple columns into meta-parameters, while collapsing multiple rows into subpopulations of cells (“clusters”). This strategy can retain key information about distinct cell subsets, while making the data more interpretable by eye. Purpose-built clustering algorithms for mass cytometry such as PhenoGraph, FlowSOM, and SCAFFOLD have emerged. A popular method to view these clusters, or drill down to view the underlying single-cell data, is to organize them by similarity on a 2D plot using t-distributed stochastic neighbor embedding (t-SNE; implemented in the popular viSNE, cytofkit, and Cytosplore packages). With this plethora of data visualization and interpretation tools, it may be difficult to decide which ones to use. New mass cytometrists should assess which tools best fit their research needs before settling on an analysis pipeline. Several academic reviews on the subject are now available to guide new users, and the online community at Cytoforum serves as an interactive and historical record of advice.
Mass cytometry analysis of mouse tumors is a key component of an ongoing NCI-funded Cancer Target Discovery and Development (CTD2) project led by Drs. Allan Balmain, Matthew Krummel, and William Weiss at UCSF. This project uses a variety of advanced technologies, including mass cytometry, to compare the immune infiltrate in immune-competent mouse models of solid tumors, including squamous cell carcinoma, breast carcinoma, and glioblastoma. These tumor models were selected because they have distinct patterns of antigen presentation and likely different strategies to evade the immune system.
Mass cytometry analysis of these tumors has revealed fine-grained subsets of different immune cell types within the tumor microenvironment and systemic effects in other immune tissues, such as lymph nodes. While much of the focus in immunotherapy has historically been on T lymphocytes, a major goal this research is to understand the diverse macrophage and dendritic cell populations within tumors, which are resolved in detail by the high-dimensional profiling of mass cytometry. Mass cytometry, and especially its application to syngeneic mouse models, is an invaluable tool for the field of systems cancer immunology. This approach of systematically comparing how the immune system behaves in the context of different tumor types and immunotherapies, across different organs and timepoints, will shed light on the underlying immune defects and inspire new approaches to restore anti-tumor immunity.
Mass cytometry complements other technologies, especially single-cell RNA sequencing, by offering rich, protein-level information on millions of cells. Over the last ten years, it has matured from a niche technology to a powerful and widely used tool, especially in the field of cancer immunology. There are now numerous review articles and online tools to help new users as they design custom-tailored antibody panels and analyze the data2,3,4. New technological tools, such as mass cytometry, coupled with appropriate mouse models of cancer, will help the field of systems cancer immunology decipher the web of cell types and responses as the immune system engages a tumor.
References
- Bendall SC, Nolan GP, Roederer M, Chattopadhyay PK. A deep profiler’s guide to cytometry. Trends Immunol. 2012; (33): 323–332. (PMID: 22476049)
- Spitzer MH, Nolan GP. Mass Cytometry. Cell. 2016; 165(4): 780-79. (PMID: 27153492)
- Olsen LR, Leipold MD, Pedersen CB, Maecker HT. The anatomy of single cell mass cytometry data. Cytometry A. 2019; 95 (2): 156-172. (PMID: 30277658)
- Mistry AM, Greenplate AR, Ihrie RA, Irish JM. Beyond the message: advantages of snapshot proteomics with single-cell mass cytometry in solid tumors. FEBS J. 2018; doi: 10.1111/febs.14730 (PMID: 30549207)