Single-Cell Metabolic Analyses of the Tumor Microenvironment: Researcher Interview with Jason Locasale
, by Peggy I. Wang
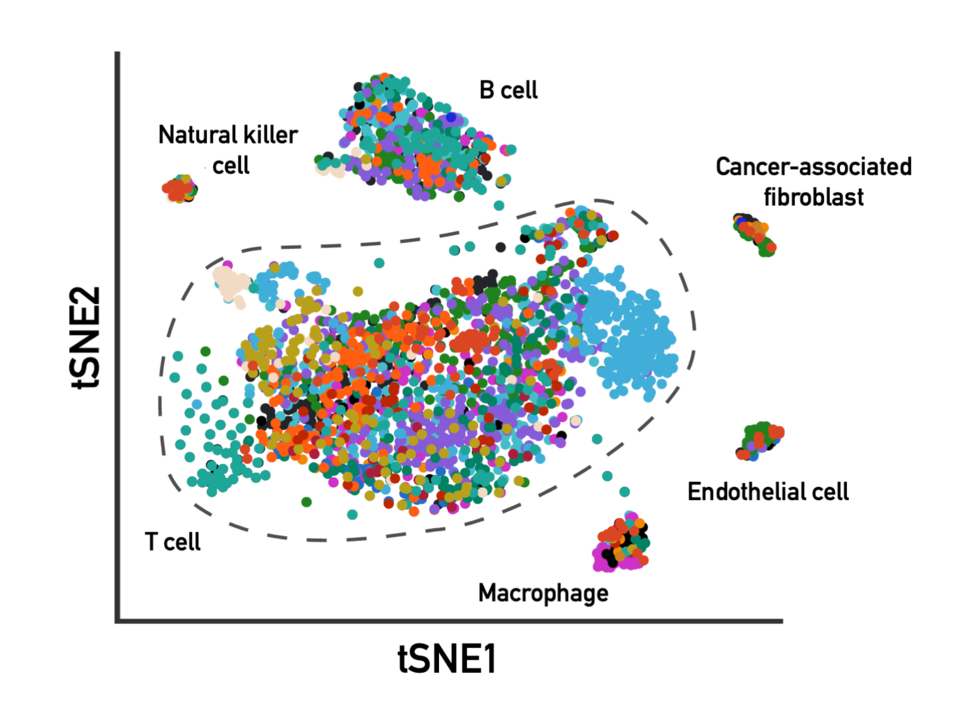
Individual immune cells from different patient tumors (indicated by different colors) have similar metabolic programs.
Credit: Adapted from Xiao et al, Nat Commun. Aug 2019 doi:10.1038/s41467-019-11738-0 CC BY 4.0
Cancer cells appear to undergo metabolic reprogramming, where the set of basic chemical reactions for converting food into energy is altered. Recent advancements in molecular technology, with single-cell sequencing in particular, are allowing researchers to bring new understanding to this century-old concept.
In a study recently published in Nature Communications, Jason Locasale, Ph.D. peeks into what is happening in the metabolism of single cells in the microenvironment of tumors. Dr. Locasale is an associate professor in the Department of Pharmacology and Cancer Biology at Duke University.
I spoke with Dr. Locasale about exploring the metabolic perspective of tumors and how it may inform future treatments of cancer.
Peggy I. Wang: How did you get into cancer metabolism?
Jason Locasale: I’ve been interested in cancer since my postdoctoral training, and our research centers around the question of how metabolic pathways influence human health, how nutrition regulates metabolism, and also how metabolism influences downstream cellular processes.
I’ve always had an interest in developing and applying technology to study these questions, both from the analytical side, for example, developing mathematical and computational tools, and the technological side, finding ways to measure the behavior of metabolic pathways. The combination of these interests really led to this study.
PIW: Is gene expression data regularly used in metabolomic studies? How did you choose the dataset you utilize for this study?
JL: Metabolomics data is typically generated with mass spectrometry, which is challenging because of the chemical diversity that we need to capture. We had been seeing single-cell data coming out and were curious about what we could learn from it. It was just a matter of digging into it and looking at what the depth of sequencing was and the quality of the data to see if we could infer something about the metabolism.
For this study, we utilized gene expression profiles of about 9,000 single cells from 19 patients with melanoma and 18 with squamous cell carcinoma of the head and neck. Both datasets were generated with full-length SMART-seq2 reads.
Luckily, metabolic genes are expressed at a high level, and we were able to modify and apply the algorithms that have been in our toolbox for a while.
PIW: So the computational methods you used were built from methods you originally developed for bulk sequencing data. What adjustments did you have to make to bring it to the single-cell realm?
JL: Actually, in the original paper, we were assembling microarray data to try to infer metabolic activity from gene expression profiles. Then when TCGA data started coming out, we had a lot of standardized mRNA measurements to work with.
Basically, we pull out the expression of the metabolic genes, group them into pathways, and make estimates about pathway activity based on the collective expression of multiple genes in the pathway. Then we also look at how each pathway interacts with another.
With single-cell data, the challenges are the noise and missing data. Fortunately, for metabolism, the genes are more abundant than things like transcription factors. There’s a disproportionate number of genes that encode for metabolic enzymes so that gives you more power to map them onto pathways.
PIW: Biologically, what did you find is going on in the metabolism of individual cells versus bulk tumors samples?
JL: Overall, we saw a lot of differences in the metabolic activity of single cells versus what has been observed from the analysis of bulk tumors. We found many novel metabolic alterations from the single-cell data.
We found something that sort of challenges some established ideas around the Warburg effect—the observation that tumors often utilize glycolysis, a kind of metabolism where they ferment carbohydrates, rather than oxidative phosphorylation, which is performed by mitochondria.
From the single-cell data, we found that when glycolysis is turned on, its accompanied by turned-on mitochondria. This goes against the idea of a metabolic switch, where you have either one or the other active. Further work is needed to resolve this discrepancy, and we can’t say for sure if the mitochondria is functioning properly even if it’s on, but it’s really interesting.
PIW: How did you determine immune cell type-specific metabolic programs, and did you find a one-to-one relationship between the type of immune cell and the type of metabolic program?
JL: We now have well-defined markers for different cell types within the tumor microenvironment. With those, we could look at all the different immune cells and look at what kind of metabolic program was running.
The metabolic signatures we measured were so robust that we could also go the other direction; we could distinguish immune cells based on their metabolic signatures.
And we’re now starting to compare these findings with the literature: for example, what’s been found in cell co-culture models. Sometimes the data agree, sometimes they don’t.
PIW: Are there limitations to inferring these metabolic measurements from transcript data?
JL: This is a major challenge of multi-omics integration. The transcripts don’t necessarily tell you the amount of protein that you have, and the amount of protein does not tell you about the function of the protein. Just because the protein level is high or low doesn’t mean that its working properly—there could be post-translational modifications or small molecules affecting the protein.
Metabolism is a whole additional layer of regulation with enzymes that have to do chemical reactions. Every metabolite is connected to every other metabolite in a connected network that’s constrained by chemical kinetics and thermodynamics. A single point mutation in a gene might rewire the whole network of enzyme reactions, or not, if a downstream step is limited.
That said, single-cell expression analysis allows us to look at enzymes at the network or pathway level, and this collective expression provides insights into what’s likely happening with metabolism. We’re a long ways from single-cell metabolomics but using the gene expression to infer metabolism is a good start.
PIW: You talk a lot about the single-cell genomics revolution. What kind of impact do you think it will have in the metabolomics space?

Dr. Jason Locasale, associate professor in the Department of Pharmacology and Cancer Biology at Duke University.
JL: I'm basically an outsider in the single-cell biology field, but every year, the amount, quality, and reproducibility of the data is just exponentially growing. We're watching this revolution in cell biology right now and I think it’s amazing.
We’re now generating single-cell data in our lab through a core and we get better data now from standard technologies than what the best data in the world was in early 2018. So it’s exciting.
There are certain questions about cancer metabolism that have always been limited by technology. It’s really hard to measure certain molecular features in a tumor. For example, in the past, the field has resorted to using cell co-culture model systems, where we try to mimic some aspect of tumor environment by growing two cell types together.
Now single-cell technologies, especially single-cell RNA sequencing, are really breaking down a lot of barriers to what we can understand about cell metabolism. We can now make direct observations of what’s happening, at least at the genomic level, and computationally infer elements of metabolism.
PIW: What are the next steps you’re excited about?
JL: This study shows that wow, single-cell data is really cool and we can open a lot of doors to understanding cancer cell metabolism in vivo. Even with this older, arguably obsolete, data set, there is enough depth and quality to gain insights into the metabolic programs of individual cells.
We’re looking forward to getting even better resolution of the tumor microenvironment at the single-cell level. This includes transcriptional and metabolic programs that sense different nutrient environments.
We’d also like to better understand “normal” biology and the metabolic programs of specific immune cells. Just as cancer cells have different metabolic features, so do specific types of immune cells, especially in their different states of activity. There is a lot of basic science left to be done in understanding immunometabolism.
We’re just beginning to see how the modulation of metabolism can influence downstream regulators of biological activity. This has a lot of implications for immunotherapy and how to potentially improve patient responses by altering metabolism with drugs or diet. For example, a recent study showed that blocking the glutamine pathway may improve T cell responses in the tumor while shutting down metabolism in cancer cells.