Cracking the Cancer Code with Computational Approaches
, by Aaron T. Griffin, M.D. Ph.D. program, Prabhjot S. Mundi, M.D., and Andrea Califano, Ph.D.
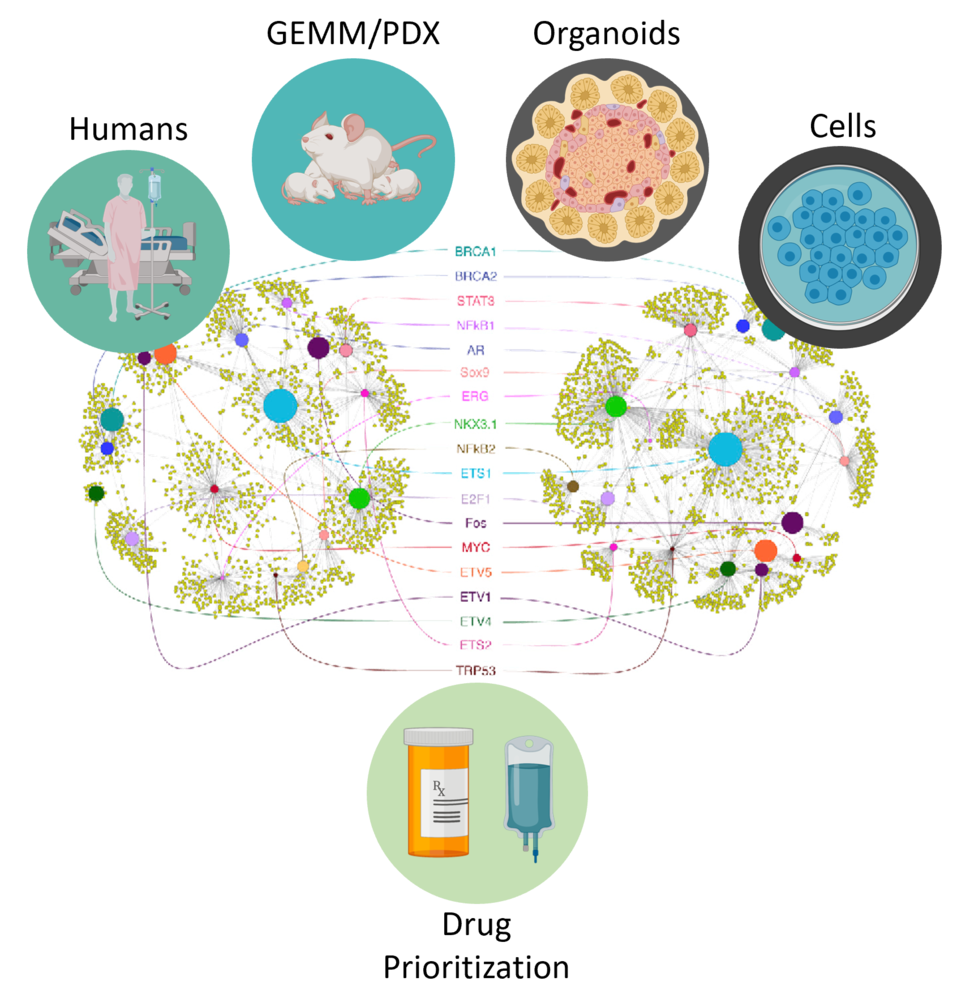
The Califano Lab employs a variety of computational approaches to help identify master regulator proteins, predict drugs that target those regulators, and test the drugs in an appropriate cancer model.
Think of the genes that make up the DNA in our cells as the musicians in an orchestra, each playing a perfectly constructed and uniquely tuned instrument, yet all silent and unheard until allowed to play their individual melodies. Think of the RNA molecules—which are ultimately responsible for producing the proteins that make up the cell—as the individual melodies that will be produced, note by note, by each musician to blend and combine into the complex symphony of the cell’s inner workings. Now think of this orchestra, much like our cells, as having more than 20,000 musicians. Clearly, no coherent piece of music could emerge spontaneously, without many synchronized conductors, each one coordinating the work of a subset of musicians, telling them when to play, when to be silent, and setting the tempo for their scores.
From this simple analogy, it should be quite obvious that, even though two orchestras may have the same musicians playing the same exact instruments, the music they produce may be vastly different, depending on their conductors and on the score and tempo they will set. Not dissimilarly, cells with virtually identical genomes in our body can act in dramatically different ways, for instance operating as a neuron, a liver cell, or a white blood cell.
To complete the picture, now imagine a few of the key instruments, such as the first violin or the piano, being badly damaged and suddenly playing dissonant notes. Chaos would ensue and a few of the conductors, disoriented by this cacophony, may even make matters worse, by over-compensating to keep the concert going at all costs, thus resulting in an even more discordant symphony. In our cells, such dystonic and uncoordinated music is the music of cancer, where mutations in several genes may end up affecting the behavior of key conductors of the cellular harmony—proteins that we have called "master regulators"—causing utter mayhem and disease.1
Until recently, the foundational assumption of precision medicine was that if you could stop the one instrument playing the most discordant music—in other words, target the most important mutated oncogene with a selective drug—then the malignant orchestra would come to a grinding halt. Unfortunately, given the large number of independent mutations that are necessary to trigger and maintain the cancer state of a human cell, targeting a single oncogene is rarely sufficient to accomplish this task and can at best slow down the “tempo” of the cell, only to see it dramatically increase again after the remaining mutations find a way to compensate for the effect of the drug. Unfortunately, this is what is being observed in the clinic with targeted therapeutics—i.e. drugs designed to inhibit specific oncogenes activated by a mutation. Indeed, on average, only 20% to 30% of patients have oncogene mutations that can be targeted with drugs and less than half respond to the therapy. More importantly, of those that initially respond, almost all will eventually relapse with a more aggressive version of the tumor that no longer responds to treatment. As a result, only 5% to 11% of patients treated with targeted therapy, on average, improve their progression free survival (PFS) and even fewer are cured.
In hindsight, considering that each cell, out of the billion that comprises a tumor mass on average, comes with its own unique set of mutations, this is not overly surprising. This is because, although targeting one mutated oncogene may kill some cells, it will not kill them all, thus resulting in the emergence of cells that are immune to the effects of the drug and will thus cause relapse. In addition, much like how some of the orchestra conductors may try to adapt the music to the sudden disappearance of one of their key instruments, using a different yet still discordant tune, tumors can dynamically reprogram themselves into a different kind of malignant state which can escape therapy by activating a new set of master regulators without requiring additional DNA mutations. For instance, prostate tumors can reprogram themselves to a very aggressive and drug-resistant neuroendocrine tumor state. Indeed, one of the reasons why some tumors are so hard to treat is that, even when effective drugs are used—e.g. against mutated oncogenes—individual cancer cells will almost inevitably find multiple ways to escape their effect.
Dr. Andrea Califano, Principal Investigator and founding chair of the Department of Systems Biology at the Columbia University Irving Medical Center, has developed computational approaches that focus on targeting the master regulator proteins responsible for directing the dissonant genetic symphony of cancer rather than the individual mutated oncogenes. The key hypothesis pursued by his lab is that, while there are billions if not quadrillions of potentially tumor-inducing DNA mutation patterns, the actual number of distinct cancer related states they can induce (i.e., those controlled by a distinct set of master regulators) is actually very small—typically 2 to 5 per tumor type. Thus, if further validated in the clinics, this would lead to the development of a much more universal set of drugs, which, rather than targeting the almost infinite number of mutated gene patterns that can start a tumor, target instead a small set of master regulator proteins.
Indeed, the lab has shown that either genetic or pharmacologic inhibition of master regulator proteins discovered by this approach can stop cancer cells dead in their tracks. To do this, the lab had to develop a variety of algorithms, which are now used in several clinical studies and trials. For instance, the Algorithm for the Reconstruction of Accurate Cellular Networks (ARACNe)2 finds the strings that connect the individual genes to master regulators that control them. Then, the Virtual Proteomics by Enriched Regulon analysis (VIPER) algorithm3 follows all of these strings to pinpoint the key master regulators of a specific tumor or even of a specific cell within a tumor. The OncoMatch algorithm then searches all available cancer models (for instance, more than a thousand cell lines that can grow in a test tube) to identify the optimal avatar for the patient’s tumor in which to evaluate the patient-relevant targets of all available FDA-approved and experimental drugs. This is achieved using large-scale perturbational assays where the RNA of the cell lines is profiled before and after perturbation with each drug. Finally, the OncoTarget and OncoTreat4 algorithms match the master regulators of a tumor to the targets of each drug to identify one or more that can reverse the activity of the master regulators and thus kill the tumor. Once considered very hard to implement, if not impossible, this strategy of targeting tumor master regulators has already been validated in numerous human malignancies.4-12 Returning to the orchestra metaphor, OncoTarget and OncoTreat identify drugs that can target the handful of out-of-control conductors of the dystonic symphony rather than the individual musicians.

Aaron T. Griffin, M.D. Ph.D. program, Prabhjot S. Mundi, M.D., and Andrea Califano, Ph.D. of Columbia University
Over three hundred FDA-approved and late-stage experimental anticancer drugs are used to perturb the optimal avatar of the patient’s tumor, as selected by OncoMatch.13,14 By using VIPER to assess the activity of the master regulators before and after the drug has been introduced in these cells, one can easily identify the drug that can target the vast majority of them. Most drugs do almost nothing in terms of inverting the activity of the master regulators and some may take down a few of them but not all. However, almost invariably, there are a few drugs that can take down the vast majority of them. In studies on 39 drugs predicted by OncoTarget and OncoTreat for patients with completely different tumors that had failed 3 to 7 lines of therapy, 60% induced a relevant response in the patient tumor transplanted into a mouse—including arresting the tumor growth or causing the tumor to shrink—and 20% caused tumor shrinkage.4 In contrast, none of the classical targeted inhibitors had any effect on these very aggressive tumors.
Interestingly, these algorithms also work on the individual cells of a tumor, thus allowing the identification of drugs capable of targeting different tumor cell subpopulations within the same tumor that would have completely different drug sensitivities. This could pave the road to a cell-by-cell approach to eliminate this terrible disease.