Brain Tumors Hack the Neuronal Microenvironment in Multiple Ways to Drive Tumor Growth
, by Cindy Kyi, Peggy I. Wang, and Subhashini Jagu
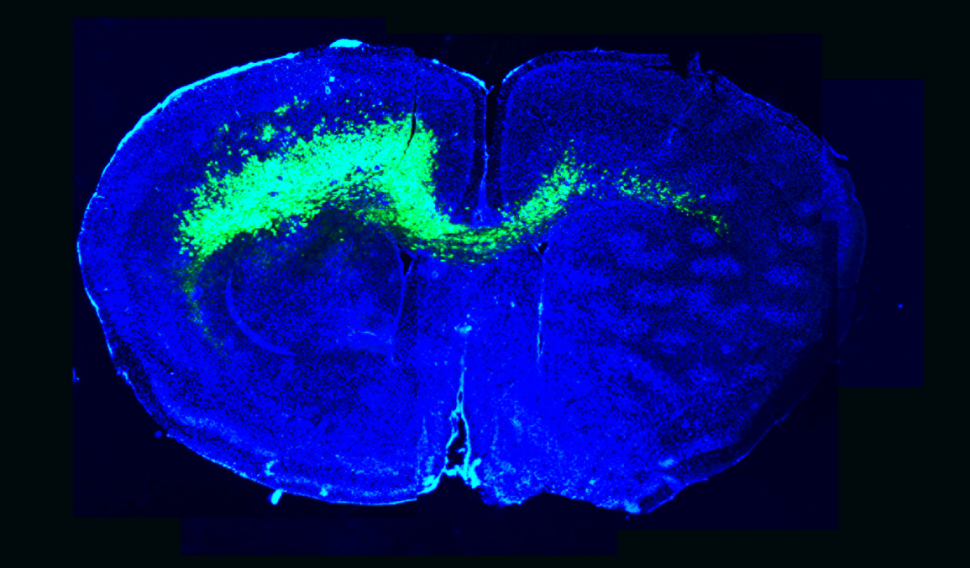
The infiltration of malignant glioma cells (green) throughout a mouse brain (blue) alters brain circuits towards hyperexcitability and sets a stage favorable for glioma growth.
Credit: Emmet Huang-Hobbs and Benjamin Deneen
This study was born out of months of in-depth discussions between me, a developmental neuroscientist, and my colleague Kenneth Scott, a visionary for functional cancer genomics. It took some time, but once we learned how to communicate across our fields, we were able to combine our technologies and build a new approach to systematically test the functions of mutations associated with a lethal form of brain cancer. Ken also taught me the meaning of team science, and our team soon grew to include Gordon Mills, a leader in precision oncology, and Jeff Noebels, world-renowned neurologist and expert on epilepsy.
Sadly, Ken passed away three years ago, while we were in the middle of screening and did not get to fully appreciate the far-reaching implications of our collaborative efforts. His vision continues to inspire all of us and if he were here today, I’m sure he would be cooking up new ways to innovate the cure for cancer.
- Benjamin Deneen
Increased neuronal activity is thought to promote the growth and spread of tumor cells in diffuse gliomas and glioblastoma, but the mechanism is unknown. This neural activity often leads to seizures—one of the few early warning signs of the deadly disease.
A recent study published in Nature examines how genetically distinct tumor subtypes exert differential effects on normal neurons, uncovering distinct molecular paths converging on the same phenotype of increased neuronal activity. The work was led by the Cancer Target Discovery and Development (CTD2) Network researchers Drs. Benjamin Deneen and Kenneth Scott and their teams at Baylor College of Medicine.
The study provides additional evidence that glioblastomas are caused by different pathways—even if the same key genes are mutated. Understanding the interactions between tumor cells and surrounding neurons in each tumor subtype driven by a particular variant will be critical for guiding new research and potential therapies.
Different PIK3CA Variants Dysregulate Synaptic Genes in Different Ways
High-throughput molecular barcoding screening technologies enable functional characterization of rare mutations. In previous work, Dr. Scott’s lab demonstrated how to apply such screens using in vitro methods to measure a mutation's ability to promote cell invasion, proliferation, and transformation, and in vivo mouse systems to measure tumorigenesis and metastasis, revealing signaling pathways driving pancreatic ductal adenocarcinoma.
PIK3CA is a core component of the RTK-RAS-PI3K pathway and mutated in 90% of glioblastomas. In this study, the researchers developed a new in vivo screening platform to identify several known and novel driver variants of PIK3CA that could accelerate tumorigenesis in a mouse model of glioblastoma and investigated the functional properties of each. The term “driver” is used here as these variants were found to accelerate tumorigenesis in glioblastoma mouse models.
Dr. Deneen and his team found that tumors driven by the PIK3CA variants exhibited varying levels of activation of the PI3K pathway, with variants C420R and H1047R showing the highest levels of activity. The researchers next focused on other downstream differences resulting from the different variants.
Expression analysis of tumors driven by these variants showed key differences in genes associated with proliferation and synapse function. For example, tumors driven by the variant C420R showed increased expression of genes associated with cell proliferation when compared to H1047R- and R88Q-driven tumors. “The stark difference in gene expression was very surprising,” says Dr. Deneen. “We knew we had to figure out how these closely related variants were behaving differently.”
The C420R or H1047R variant driven tumors dysregulated different subgroups of synaptic genes. Through antibody staining of mouse tumors, the researchers found an increase in excitatory synapses and a decrease in inhibitory synapses, a clue for how these particular variants may lead to synaptic imbalance.
The Genetic and Cellular Context Further Modulate Effects of Specific Variants
Glioblastoma mice bearing tumors accelerated by C420R and H1047R variants showed a very early onset of biological neural network hyperexcitability and seizures, whereas tumors accelerated by R88Q variant showed a delayed onset of seizures or changes in network activity. Experiments in patient-derived xenografts (PDXs) further demonstrated that tumors accelerated by these different variants of PIK3CA exhibit distinct physiological properties, including the presence and timing of hyperexcitability and seizures.
Expression of the variants in non-glioblastoma model mice further highlighted their differences. While the expression of H1047R in non-glioblastoma model mice still led to hyperexcitability and seizures, the same was not true for R88Q or C420R.
The researchers then tested if either the C420R or H1047R PIK3CA allele overexpressed in glial cells causes neural hyperexcitability in neighboring wild-type neural cells. Using a cell co-culture system, they found that C420R expressed in glial cells promotes synapse formation between neurons, indicating that C420R somehow influences neighboring cells. This so-called “cell non-autonomous” behavior was not found for H1047R.
A Novel Model for Manipulating Neurons to Promote Tumorigenesis
To explore how cells with C420R exert an effect on neurons, the researchers looked for secreted genes known to play a role in synapse formation. Analysis of gene expression data identified glypican 3 (GPC3), which was upregulated not only among C420R-driven tumor mouse models but also in human glioblastoma samples.
Experiments in their mouse glioblastoma model showed that GPC3 plays a key role in promoting abnormal synapse formations and results in an earlier onset of seizures and hyperexcitability. Dr. Deneen’s team found that overexpression of GPC3 accelerates tumorigenesis independently of C420R.
Through further characterization of GPC3, the researchers describe a novel mechanism in which secreted proteins from gliomas stimulates neuronal activity by triggering synaptogenesis during early tumor formation, working together with neurons to orchestrate both network hyperexcitability and growth.
This work demonstrates a myriad of ways glioblastoma cells can interact with, and possibly manipulate, neighboring neurons. “Though these variants we investigated differ only by a single amino acid, they can lead to fundamentally different tumors,” says Dr. Deneen. “It really speaks to the heterogeneity that we see in cancer and highlights the importance of carefully examining each variant to make progress in precision medicine for this incredibly lethal disease.”