Glutamine, Cystine, and Cancer Susceptibilities
, by Jim Hartley
Cancer cells grow rapidly and generate abundant reactive oxygen species (ROS) in doing so. To counter these toxic entities cancers typically upregulate a variety of antioxidant systems, including increasing the amount of glutathione (GSH), to the point that ROS may be lower in tumors than in adjacent normal tissue.1 GSH is the most abundant metabolite in cells, and is synthesized from three amino acids, glutamine, cysteine, and glycine. Consequently a variety of approaches are being tested to decrease the capacity of cancer cells to protect themselves from ROS. Four recent papers illustrate the increased sophistication of our knowledge of ROS control and glutamine and cysteine metabolism in cancers.
- Cramer, et al.2 noted cancer's high requirement for cysteine for GSH synthesis, and designed an enzyme that degrades both cysteine (CSH) and its disulfide form cystine (CSSC) in the blood. They introduced two mutations to increase cyst(e)inase's activity on both CSH and CSSC, and PEGylated it to increase its in vivo half life. When injected into mice the enzyme decreased the growth of prostate cancer allografts, and increased survival in a mouse model of human chronic lymphocytic leukemia. Of particular note is the possibility that cyst(e)inase would interrupt contributions of CSH from the tumor stroma, as has been reported in human CLL patients.
-
Muir, et al.3 discovered that the dependence of some cancer cell lines on glutamine is artifactual. Historically, cultured cancer cells consume far more glutamine than any other amino acid, and in fact inhibition of glutaminase with the drug candidate CB-839 has been tested in human clinical trials, thus far without benefit to patients. Elegant experiments revealed that much of the glutamine dependence of cancer cell lines is driven by the superphysiologic concentration of cystine in standard growth media: in these cells high cystine drives the cystine/glutamate antiporter xCT to import cystine and excrete glutamate, and the cells must then import glutamine to maintain intracellular glutamate. Such cells are more sensitive to CB-839 , and cells with lower xCT expression are less sensitive. And in contrast to Cramer, et al., who sought to decrease blood cystine levels, Muir, et al. showed that injections of cystine into tumor-bearing mice increased uptake of glutamine in tumors, potentially making them more sensitive to glutaminase inhibitors.
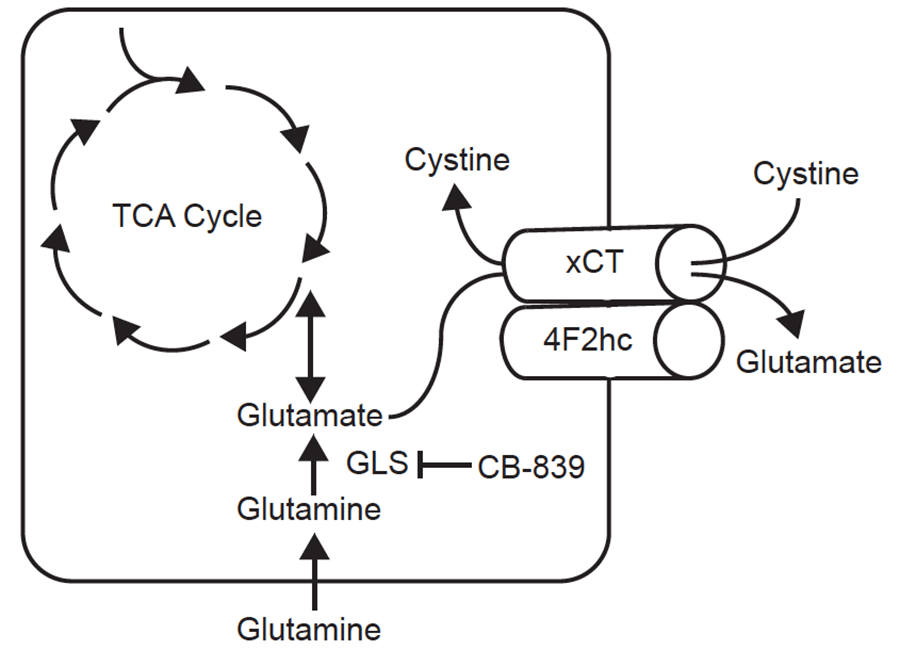
Sketch of glutamine and cystine metabolism, from Muir, et al.3
- Sayin, et al.4 explored the control of glutamine/glutamate metabolism in cancer cells. NRF2 in the master transcription factor that controls responses to ROS, and KEAP1 protein binds to and downregulates NRF2. When ROS increases KEAP1 is oxidized and releases NRF2 so that its transcriptional programs are upregulated. Among the NRF2 targets is the glutamate/cystine antiporter, xCT. Cells that are defective in KEAP1 have increased NRF2 transcription and increased import of cystine and export of glutamate, thus requiring more import of glutamine from the milieu to regenerate intracellular glutamate via glutaminase activity. Accordingly, such cells are sensitive to the glutaminase inhibitor CB-839, but are rescued from CB-839 toxicity by either lowering extracellular cystine or raising extracellular glutamate. However, Sayin, et al. suggest that CB-839 toxicity in these cells is not due to lack of GSH for buffering ROS, but instead is caused by lack of glutamate inputs through a-ketoglutarate into the TCA cycle.
- Finally, Romero, et al.5 show that consistent with the results of Sayin, et al., xenografts of human lung tumors are more sensitive to glutaminase inhibition if they have KEAP1 mutations.
Together these papers provide an important update to our understanding of how cells change their metabolism when dysfunctional growth causes reactive oxygen to threaten their survival, and how those changes might influence therapeutic interventions.