Enzymology of GTP-competitive RAS inhibitors
, by Ken Westover

Dr. Ken Westover.
The belief that the active site of RAS cannot be drugged is easy to find throughout the literature, but the experimental, or even quantitative theoretical evidence supporting this belief is not as easy to find. I was not working in the field when this view first emerged but I suppose it is based on enzymological arguments. Enzymology provides a theoretical basis for understanding the behavior and limits of catalytic macromolecules and is a vital tool for drug discovery. Enzymological researchers often employ computer-aided simulations to both model experimental data and make predictions. At their core, simulations are composed of relatively simple calculations which take into account the concentrations of components (enzyme, ligands and substrates) and rate constants describing how quickly binding events or chemical conversions occur. However, when multiple transformations are linked together in a multi-component reaction scheme, the mathematics become more complicated and computers provide valuable assistance in crunching the numbers. An elementary scheme for the interaction of KRAS with a GTP competitive inhibitor is shown in Figure 1A. Running a simulation indeed predicts that reversible inhibitors (Kinact = 0) are unlikely to be effective competitors when matched up against the picomolar affinity between RAS and GTP1-3, especially given that cellular concentrations of GTP are in the hundreds of micromolar range4-14. Achieving significant RAS inhibition with reversible inhibitors would require compounds with low picomolar dissociation constants (see Figure 1B), a formidable and likely impossible challenge. While this may be true for reversible inhibitors, what about covalent inhibitors? What does enzymology predict for a covalent inhibitor ‘hitting’ the active site of KRAS?
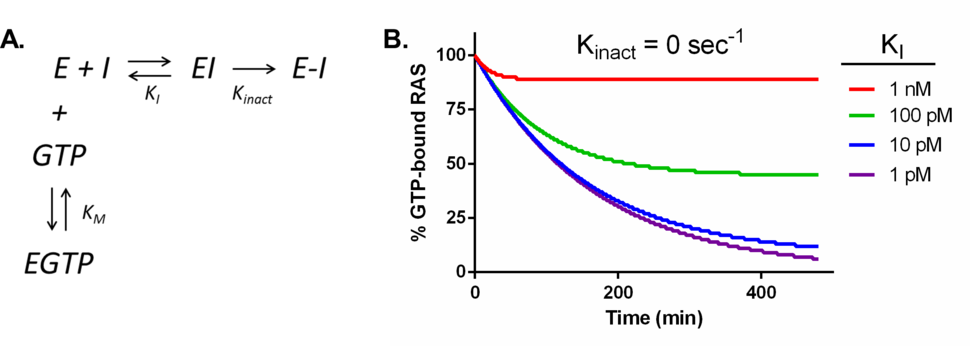
Figure 1. Kinetic modeling KRAS inhibition by reversible inhibitors. (A) Schematic of the model system where E = RAS, I = inhibitor, EI = a transient complex of inhibitor and RAS, E-I = a covalent complex between KRAS and inhibitor, EGTP = a transient complex between RAS and GTP, KM = the dissociation constant for GTP, KI = the inhibition constant for inhibitor and Kinact = the rate constant for covalent inactivation of KRAS. (B) Simulated inhibitions for reversible inhibitors with Ki between 1 pM and 1 nM. Simulation conditions are as follows: KRAS = 400 nM22, GTP = 200 µM4-14, Inhibitor = 50 µM, KM = 500 pM1-3. Simulation was done using Gepasi23.
This is a unique time in the history of drug development when it comes to covalent inhibitors. With the recent emergence of FDA approved examples of targeted, covalent drugs like Afatinib and Ibrutinib, covalent inhibitors have seen something of a renaissance in recent years15-16. Indeed, covalent inhibitors are not new; there are plenty of effective examples such as aspirin, penicillin and omeprazole17. What is new is the emergence of efficient techniques and the collective experience within the medicinal chemistry community that has enabled the rational design of multiple examples of covalent, targeted agents18. When viewed from the perspective that cellular signaling often uses covalent modifications to drive biology, it doesn’t require a big leap to hypothesize that covalent inhibitors may provide unique pharmacological advantages if reactivity can be adequately controlled. So what benefits would covalent chemistry provide for drugging the active site of KRAS?
Although most people reading this probably already know many details about RAS structure, it seems worth mentioning in this context that the epicenter of RAS’s influence is the active site. To be more precise, it is the structural consequences of RAS binding to either GTP or GDP that differentiates between RAS signaling or not. It is therefore not surprising that the observable structurally dynamic parts of the RAS core are limited to a few loops and helices immediately adjacent to the active site, and that the structural overlap where most RAS effectors converge in their interactions with RAS comprises these same elements: the switches (see Figure 2). Though targeting the active site was dismissed early on as a viable strategy, as David Heimbrook recently discussed, it is hard to conceive of a better place a priori to put small molecules if one wishes to disrupt RAS signaling.
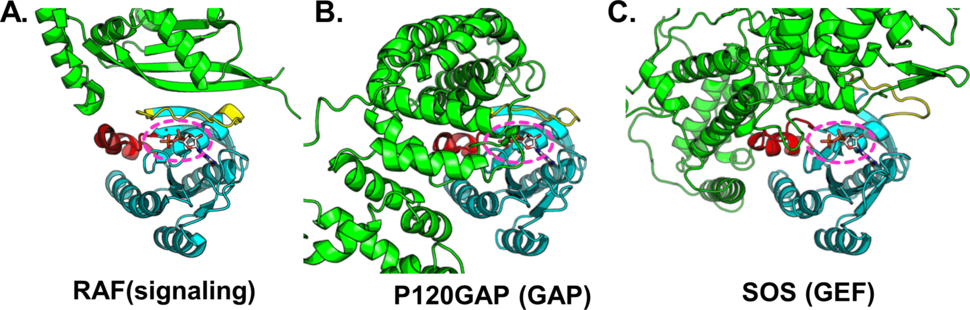
Figure 2. Location of effector, GAP and GEF binding on RAS. X-ray crystal structures for RAS in complex with (A) RAF (3DDC), (B) p120GAP (1WQ1) and (C) SOS (1BKD) are shown. Each constitutes a distinct class of RAS-interacting protein (Raf – signaling, p120GAP – nucleotide hydrolysis, SOS – nucleotide exchange) but all interact with the RAS switches (colored yellow and red). For all complexes RAS has been superimposed for comparable orientation between panels. RAS is cyan and interacting protein is in green. GDP is shown as sticks and is highlighted with a pink dotted line.
The activating G12C mutation in KRAS is an intriguing foothold for getting small molecules into the active site using covalent chemistry. Among amino acids cysteine is distinct because it contains a highly nucleophilic thiol group, a common target for the electrophilic warheads of many targeted covalent compounds. Furthermore, the KRAS G12C isoform is not an insignificant drug target; it is the most common KRAS mutation in non-small cell lung cancer, presenting in approximately 25,000 new cancer cases annually in the US. If the G12C mutation could be effectively exploited for covalent chemistry, this would not only provide a way to get inhibitors into the active site, but also a powerful mechanism for achieving compound selectivity. So what does theoretical enzymology predict?
Another simulation provides an interesting result. An inhibitor with moderate reactivity (Kinact = 0.1 sec-1) and a relatively potent, but plausibly achievable, inhibition constant (modeled between 1 and 100 nM) (ref. 19) would decrease the concentration of activated Ras substantially within a few hours (Figure 3).
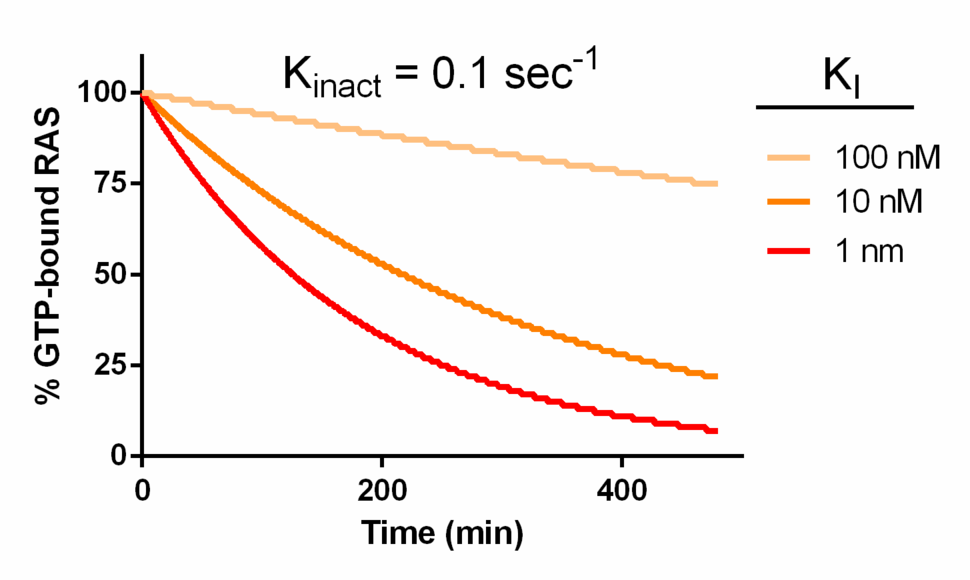
Figure 3. Kinetic modeling KRAS inhibition by covalent inhibitors. Assumptions were identical to Figure 1, except where noted.
Granted, this is a simple simulation and does not take into account many factors that may be important in a cell, but as a first estimation it provides some encouragement. Further encouragement comes from proof-of-concept experiments done with a ‘tool’ covalent GDP-analogue compound our collaborative group developed, SML-8-73-1, which showed that covalent chemistry enables irreversible entry of a synthetic small molecule inhibitor into the RAS active site and biochemical inactivation of KRAS, even in the face of supraphysiological levels of GTP and GDP20, 21. SML-8-73-1 is not cell permeable, limiting biological study. However, if the covalent approach can be adapted to compounds that have better pharmacological properties than the GDP tool compound, they may allow us to answer important questions that biochemical experiments and simulations cannot. Chief among these will be – how much RAS inactivation is required to see biological effects in various contexts? We’re working to develop such compounds, but in the meantime I can’t help but ask the following question: If it was enzymology that convinced the field that drugging the active site of KRAS was not possible, will enzymology now persuade a few people that, at least for KRAS G12C, the KRAS active site can be drugged?
About the Author
Kenneth Westover obtained his MD and PhD degrees at Stanford, where his research mentor was Roger Kornberg. He combined his residency in radiation oncology at Harvard with work in the laboratory of Nathanael Gray and contributed to the discovery of a covalent inhibitor of KRAS G12C. Dr. Westover moved to UT Southwestern in 2012.
References
- Feuerstein J, Goody RS, Wittinghofer A. J Biol Chem 1987; 262: 8455-58.
- Finkel T, Der CJ, Cooper GM. Cell 1984; 37: 151-8.
- Hattori S, et al. Mol Cell Biol 1985; 5: 1449-55.
- Werner A, et al. J Chromat B 1987; 421: 257-65.
- Fürst W, Hallström S. J Chromat B 1992; 578: 39-44.
- Pilz R, Willis R, Boss G. J Biol Chem 1984; 259: 2927-35.
- Hauschka PV. Meth Cell Biol 1973; 7: 361-462.
- Snyder FF, Cruikshank MK, Seegmiller JE. Biochim Biophys Acta 1978; 543: 556-69.
- Jackson RC, et al. Life Sciences 1976; 19: 1531-6.
- Weber G, et al. Adv Enzyme Reg 1985; 23: 81-99.
- Keppler DO, Pausch J, Decker K. J Biol Chem 1974; 249: 211-6.
- de Korte D, et al. Anal Biochem 1985; 147: 197-209.
- Wright DG. Blood 1987; 69: 334-7.
- Pilz RB, et al. Cell Growth Diff 1997; 8: 53-9.
- Byrd JC, et al. New Engl J Med 2013; 369: 32-42.
- Miller VA, et al. Lancet Oncol 2012; 13: 528-38.
- Singh J, et al. Nature Rev Drug Discov 2011; 10: 307-17.
- Liu Q, et al. Chem Biol 2013; 20: 146-59.
- Noe MC, Gilbert AM. Ann Rep Med Chem 2012; 47: 413-39.
- Hunter JC, et al. PNAS 2014; 111: 8895-900.
- Lim SM, et al. Angew Chem 2014; 53: 199-204.
- Fujioka A, et al. J Biol Chem 2006; 281: 8917-26.
- Mendes P. Comput Appl Biosci 1993; 9: 563-71.