Don't Miss the Connection: The Role of RAF1 and ROKα in RAS-Driven Cancers
, by Andrea Varga and Manuela Baccarini

Andrea Varga, Ph.D. (L) and Manuela Baccarini, Ph.D. (R)
A recent paper by the Barbacid lab has corroborated the relevance of the kinase-independent function of RAF1 (aka CRAF) in RAS-driven tumors of epithelial origin. Barbacid and colleagues1 show that inactivation of the RAF1 gene in a model of sporadic KRAS- or KRAS/p53-induced lung cancer causes tumor regression without major toxicities, the latter probably due to the fact that ERK signaling is not impacted by RAF1 deletion. In line with this, a kinase-dead RAF1 mutant rescues the phenotype, suggesting that efforts to target RAF1 in tumors should aim at decreasing expression rather than at blocking kinase activity.
This demonstration of a kinase-independent role of RAF1 in lung cancer is the most recent in a series of experiments showing that RAF1 is essential for the regulation of ERK-independent signaling. Specifically, RAF1 is heavily involved in pathway cross-talk mediated by physical interaction with other proteins2, namely the proapoptotic kinases ASK1 and MST2 and the cytoskeleton-based kinase ROKα (the product of the ROCK2 gene). The activation of these pathways caused by RAF1 deletion is responsible for the phenotypes of RAF1-deficient cells, tissues, or animals. In particular, ROKα activation is behind many of the phenotypes caused by RAF1 ablation, including defects in migration and apoptosis, which can be reverted by treatment with ROK inhibitors or by the expression of a kinase-dead ROKα3,4, and in the collective migration of endothelial cells required for tumor-induced sprouting angiogenesis5. In the context of RAS-driven epidermal tumorigenesis, ROKα-induced keratinocyte differentiation6 is responsible for the lack of tumor development in RAF1-deficient epidermis, as shown by the reversal of the phenotype achieved by topical treatment with a ROK inhibitor7. In all these instances, complexes between RAF1 and ROKα, promoted by activated RAS, could be identified; and more recently ROKα was shown to interact specifically with a phosphorylated RAF1 species generated in the context of RAS-induced RAF dimerization8. This is in contrast to the interaction with the proapoptotic kinase MST2, which occurs selectively with inactive RAF1 (ref. 9). Thus, the picture that emerges is that RAF1 operates as a signaling hub promoting survival by inhibiting MST2, and integrating and possibly timing different outputs (proliferation, invasion, differentiation) via ERK activation and ROKα inhibition downstream of activated RAS (Figure 1). Importantly, RAF1 does not bind to the ROK paralog ROKβ (aka ROCK1).
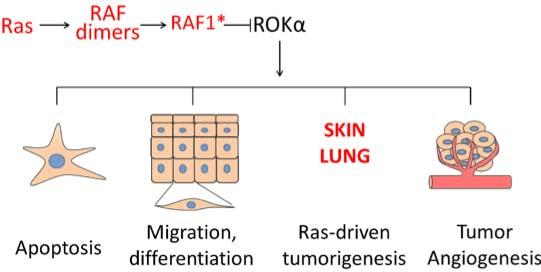
Activated RAS (and other small G proteins such as RAP1) promote the formation of a phosphorylated RAF1 species that interacts with ROKα and dims its activity, allowing the regulation of apoptosis, migration, differentiation which is essential for tumor development, maintenance, and sprouting angiogenesis.
The RAS/RAF1/ROKα connection is essential for the development and maintenance of tumors in the epidermis and in the lung, but it has not been investigated in depth in other tumors in which KRAS is frequently activated, such as colorectal or pancreatic cancer. For colorectal cancer, studies in isogenic cell lines have shown that KRAS uses RAF1 to inhibit ROK signaling and increase invasion/metastasis10; in addition, RAF1 silencing or inhibition promoted differentiation of colorectal cancer cells derived from three different donors, reducing their clonogenicity and tumorigenicity11. In both studies, the role of RAF1 was MEK-independent. In contrast, RAF1 was found to be dispensable in the only published study analyzing its function in a model of KRAS-driven pancreatic cancer12.
For those who might consider adding the ROKs to their research program, there are a few nuances that should be borne in mind.
- ROKα and its paralog ROKβ share a conserved kinase domain (87% amino acid identity), while the C-terminal regulatory domain shows less homology13. Gain-of-function experiments can be conducted using deletion of the regulatory domain to generate a constitutively active kinase14, which however loses paralog-specific function. Conditional expression of this construct in vivo has been used, for instance, to correlate high ROK activity with increased proliferation and migration of tumor cells as well as with collagen deposition and tissue stiffness15.
- Paralog-specific ROK antibodies targeting the C-terminal regulatory domains are available from BD Biosciences, while the ROK antibody available from EMD-Millipore, which targets the kinase domain of ROK, will recognize both isoforms. ROK isoforms phosphorylate the same substrates, proteins responsible for stress fiber formation and actomyosin contraction (MLC, MYPT), for the stabilization of the actin cytoskeleton (cofilin) and for the connection between the plasma membrane and the actin cytoskeleton (ezrin). Phosphospecific antibodies to these targets exist and ROK activation can be detected in lysates by immunoblotting, in cells and tissues by immunofluorescence, or by immunohistochemistry in paraffin-embedded samples, implying that they could be used as biomarkers.
- These readouts will indicate ROK activation but will not discriminate between the two ROK paralogs. This distinction has long been deemed irrelevant because of the high degree of conservation of the ROK kinase domain. However, loss of function studies have shown that discriminating between paralogs is crucial for interpretation. Germline knockout of ROKα or ROKβ has demonstrated that one paralog cannot compensate for the loss of the other during embryonic development16,17. The paralogs have been shown to be expressed at different levels and to be differentially regulated in fibroblasts (ROKα binds phosphatidylinositol 3,4,5P(3) and is sensitive to its levels), and RNAi has shown that they regulate different cytoskeletal structures in these cells18. In keratinocytes, ROKα, but not ROKβ, induces differentiation6, and the two paralogs have distinct roles in the assembly and turnover of focal adhesion complexes19. Thus, to dissect the role of ROK paralogs in a given system, it is necessary to knock down the paralogs using RNAi, or perform CRISPR-Cas9 knockouts as in ref. 20. Alternatively, paralog-specific dominant negative mutants (kinase-dead mutants of full length ROK or the C-terminal, regulatory domain of ROK) have been used to demonstrate the involvement of a specific isoform3,4,14,20,21.
- Silencing or the expression of dominant negative mutants are not practicable in vivo; therefore, one has to resort to the use of chemical inhibitors. The two most used inhibitors are the ATP-competitive compounds Y-27632 (mostly in vitro) and fasudil, approved for human use as a vasodilator in Japan and China (in vivo). These inhibitors have a relatively low binding affinity and can inhibit other kinases; therefore, more potent and more isoform-specific inhibitors are being developed22. This is certainly justified by the preclinical studies in the literature, which show that the effect of ROK inhibitors is context specific. ROK inhibition promotes tumor growth in the otherwise refractory RAF1-deficient epidermis7 and in the forestomach epithelium, particularly when used in combination with RAF inhibitors23; this is due to the inhibition of ROKα activity, which promotes keratinocyte differentiation in the absence of RAF1. In contrast, the same inhibitor reduced tumor growth in an experimental model of KRAS-driven non-small cell lung cancer (NSCLC)24, similar to the one used by Sanclemente et al. when used in combination with the proteasome inhibitor bortezomib. The molecular basis for this is that both ROKβ and the proteasome are upregulated in that model, as a result of the activation of the transcription factor GATA2 which was shown to be required for the survival of NSCLC24.
The existing ROK inhibitors have the great advantage that they are safe for use in humans. However, their overall effect will often depend on their combination with other inhibitors targeting pathways activated in parallel, as for example in hematopoietic malignancies (reviewed in ref. 13). Only a thorough understanding of each specific experimental system will determine whether inhibitors targeting both paralogs, or a specific isoform, will have a beneficial or a detrimental effect on cancers driven by RAS.
About the Authors
Andrea Varga studied the RAF1/ROKα interaction in the Baccarini lab, and is now in the Department of Biophysics of the Semmelweis University in Budapest, where she investigates connections between RAF signaling and the mechanobiology of endothelial cells. She can be contacted via email at matkovicsne.andrea@med.semmelweis-univ.hu.
Manuela Baccarini is Professor of Cell Signaling at the Max F. Perutz Laboratories, University of Vienna, where she also coordinates the PhD program “Signaling Mechanisms in Cellular Homeostasis” and the Vienna Doctoral School “Molecules of Life”. She can be contacted via email at manuela.baccarini@univie.ac.at.