Tissue Health Prevails: KRAS Mutant Cells are Outcompeted from Adult Pancreas Tissues

William Hill, Ph.D., and Catherine Hogan Ph.D.
This research was carried out at the European Cancer Stem Cell Research Institute, School of Biosciences, Cardiff University in UK where Catherine Hogan is Lecturer and Group Leader. This work was part of William Hill’s PhD thesis in Dr Hogan’s lab, developed with the support of co-authors. This project was launched with a Pancreatic Cancer UK Research Innovation Fund and supported by MRC PhD studentship and Amser Justin Time funding (Pancreatic cancer charity based in Wales, UK).
Most human cancers start with epithelial cells that have acquired genetic mutations while surrounded by healthy neighbours. Genomic profiling of histologically normal tissue from a range of body sites has revealed that as we age our tissues collect genetically mutant cells, yet cancer occurrence remains a relatively rare event. The primary function of an epithelial tissue is to act as a protective barrier to the external environment. Unsurprisingly, several processes have evolved that maintain correct tissue organisation and balance the numbers of cells that contribute to the barrier. How genetically mutant cells expand and grow to form a tumour within this tightly controlled environment remains poorly detailed. Equally, we lack a clear understanding of how epithelial tissues maintain homeostasis in adulthood, particularly when tissues accumulate aberrant cells.
A plethora of research now supports the principle that cells carrying cancer-causing mutations compete for space and survival in tissues1,2. Cell competition is an evolutionarily conserved process that eliminates cells from tissues3 and is triggered by genetic mutations, relative differences in growth rates, or mechanical properties of cells4,5. Importantly, competitive interactions occur when cells differ in ‘cell fitness’ to their neighbours, resulting in the elimination of the ‘loser’ cell from the tissue. For this reason, cell competition is viewed as a quality control mechanism that ensures only the best cells contribute to tissue function.
Cells expressing oncogenic HRAS G12V are often eliminated from epithelial tissues via apical extrusion6,7, segregation8 or dynamic tissue remodelling9, suggesting that competitive interactions with wild type neighbours are tumour preventative. The mechanisms underlying how RAS mutant cells are detected in tissues and subsequently eliminated, and the relevance of competition to human cancer have remained important open questions.
Human pancreatic ductal adenocarcinoma (PDAC) starts sporadically with cells that have acquired genetic mutations in oncogenic RAS, with KRAS G12D being the most prevalent driver mutation. The adult pancreas is divided into two main tissue compartments based on function: the exocrine acinar cells produce digestive enzymes that are secreted and carried to the duodenum via the ductal epithelial network. The endocrine islet cells produce and secrete insulin to regulate blood glucose. Unlike the skin or intestine, which replenish over a period of days, the adult pancreas is a slow growing tissue and uses cell reprogramming pathways (plasticity) to regenerate following injury. How the pancreas maintains tissue health following mutational insult is less well understood.
Using the genetically engineered KC mouse model (Pdx-1 CreER LSL-KrasG12D/+) of pancreatic cancer, we set out to model sporadic tumorigenesis in adult pancreas and investigate whether competitive interactions between KRAS G12D-expressing and normal cells prevent or promote early tumorigenesis10. The mouse model uses a tamoxifen inducible Cre recombinase and loxP technology to drive expression of KRAS G12D and exploits a red fluorescent protein (RFP) reporter to label mutant cells in both exocrine and endocrine compartments in adult tissues. By administering a single low dose of tamoxifen to adult mice, we induce RFP and KRAS G12D expression in low numbers of cells in an otherwise healthy epithelium, simulating sporadic tumorigenesis.
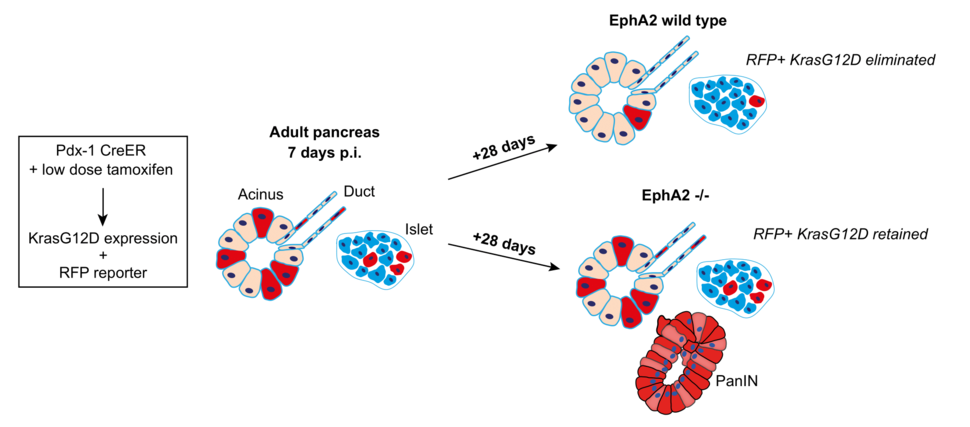
KRAS G12D cells are outcompeted from adult pancreas tissue compartments in an EphA2-dependent manner. We use the KC mouse (Pdx-1 CreER, LSL-KrasG12D; ROSA26 LSL-tdRFP) to model sporadic tumorigenesis in pancreas tissues. By administering a single low dose tamoxifen to adult mice, we induce KRAS G12D (and RFP reporter) in low numbers of cells, surrounded by healthy neighbours. At 7 days post induction (p.i.), tissues contain ~20% of RFP-labelled cells. At 28 days later, all compartments of the pancreas (exocrine, ductal epithelial, endocrine) have cleared RFP+ KRAS G12D cells. KRAS G12D cells are retained in EphA2-/- tissues and PanIN lesions are more frequent in tissues at this time point
We combined quantitative imaging approaches and qPCR of genomic DNA to measure the amount of RFP in tissues over time. We found that low dose tamoxifen induced recombination in ~25% of the tissue and this remained stable in KRAS wild type tissues over a period of 5 weeks. In contrast, the amount of RFP significantly decreased in KRAS G12D tissues, suggesting that RFP-labelled KRAS G12D cells are lost from tissues over time. Scoring RFP+ cells in each of the epithelial compartments, we found that ductal epithelial compartments and endocrine islets have significantly fewer RFP-labelled KRAS G12D cells compared to wild type controls. Importantly, we observed no change in RFP levels in KRAS G12D tissues treated with high dose tamoxifen. In this experiment, recombination occurs in ~75% of the tissue, thus the proportion of normal cells in tissues is significantly decreased. We conclude that when present in tissues in low numbers, KRAS G12D cells are eliminated, and this requires the presence of normal cells. Considering exocrine acinar cells constitute ~80% of the pancreas, our data suggest that KRAS G12D cells are outcompeted from all tissue compartments in adult pancreas.
What is the mechanism underlying KRAS G12D cell elimination in the pancreas? We first monitored changes in cell death and proliferation in tissues. We found that the rate of cell proliferation and cell death in the adult pancreas is very low, regardless of genotype, and we found no evidence of apoptosis of KRAS G12D-expressing cells, suggesting that competition does not trigger KRAS G12D cells to die. Moreover, we found no evidence that normal cells have a growth advantage over mutant cells.
We have previously shown that EphA2 receptor (of the Eph-ephrin family of cell-cell communication signals) drives the segregation and extrusion of RAS G12V-expressing cells from normal cells in simple epithelia8,11. Functional DEph is also required to eliminate RAS G12V cells from the wing imaginal disc in Drosophila melanogaster in vivo8, suggesting that Eph receptors have an evolutionarily conserved role in RAS mutant-normal cell-cell communication. To address whether EphA2 is required in mammalian tissues in vivo, we crossed the KC mouse onto an EphA2 knockout mouse12 to generate Pdx-1 CreER LSL-KRAS G12D/+; Rosa26 LSL-tdRFP; EphA2-/-10. We found that loss of functional EphA2 had no significant effect on cell proliferation in tissue; however, apoptotic events were significantly higher (albeit at low levels) in young EphA2 -/- tissues. An intriguing possibility is that EphA2 may be required to efficiently extrude dying cells from tissues13. Remarkably, we found that RFP-labelled KRAS G12D cells are retained in exocrine and endocrine compartments in EphA2-/- tissues. Thus, EphA2 is required to drive the elimination of KRAS G12D cells from adult pancreas epithelia.
Based on our previous work8, we reasoned that KRAS G12D cells are triggered to segregate and adopt a contractile morphology when surrounded by normal cells. Using immunofluorescence tomography14 and 3D imaging, we measured changes in the shape and size of RFP-labelled cells in tissues fixed at early time points, prior to when cells are cleared from tissues. Consistent with our previous findings, we found that KRAS G12D cells form tightly packed round clusters in tissues, suggesting mutant cells segregate from normal neighbours. Interestingly, we showed that KRAS G12D cells significantly decrease in cell volume, while normal cells directly neighbouring mutant cells significantly increase in cell volume, suggesting that normal cells expand in size to compensate for the loss of mutant cell mass. We also found that E-cadherin significantly decreased at cell-cell contacts between KRAS G12D and normal cells in both exocrine and endocrine compartments, supporting the idea that KRAS G12D cells segregate away from normal cells. Changes in cell volume, cell clustering and E-cadherin localisation were not observed in EphA2-/- tissues. In fact, E-cadherin significantly increased at cell-cell contacts in EphA2-/- tissues, suggesting an autonomous role of EphA2 in the regulation of E-cadherin turnover. Together our data suggest that pancreatic tissues remodel cell-cell adhesions and cell shape/volume to remove KRAS G12D cells and this dynamic remodelling requires functional EphA2.
How are KRAS G12D cells eliminated from the tissue? Using imaging and mathematical modelling approaches, our data suggest that mutant cells are outcompeted at the boundary with normal cells, and that clusters of KRAS G12D cells progressively lose cells over time, irrespective of cluster size. Interestingly, KRAS G12D cells are lost from ductal epithelial compartments by 7-days (post tamoxifen induction), whereas acinar and islet compartments eliminate cells over 35-days, suggesting that different processes may be required to remove KRAS G12D cells depending on tissue location. Further work is needed to determine the precise mechanism by which KRAS G12D cells are removed from tissue compartments. Indeed, local tissue architecture and/or differences in EphA2/ephrin ligand expression are likely factors to consider.
Finally, we asked whether cell competition is relevant to tissue health and disease. Human PDAC predominantly expands from premalignant lesions termed pancreatic intraepithelial neoplasias (PanINs), which arise from KRAS G12D cells; however, an additional insult (e.g., inflammation) is required to drive PDAC. We found rare PanINs in aged KRAS G12D tissues, suggesting the population of KRAS G12D cells that are never eliminated from tissues may be cancer cells of origin. Remarkably, PanINs appeared in KRAS G12D EphA2-/- tissues at early time points and with increased frequency. We conclude that loss of functional EphA2 leads to an increase in KRAS G12D cells retained in tissues, thereby increasing the frequency of oncogenic niches, and promoting disease. Therefore, competitive interactions with normal cells promotes tissue health and prevents disease initiation.
Considering KRAS G12D mutations drive all stages of pancreatic cancer, our research indicates that KRAS G12D cells must first override cell competition signals to remain in tissues and drive early disease. Understanding how KRAS G12D cells evade competition with normal cells to become ‘winner’ cells is an exciting line of investigation and is our current focus. Indeed, a better understanding of how tissues maintain healthy aging and the events underlying early disease may improve our ability to detect pancreatic cancer at early time points and improve patient prognosis.
For further information, please contact Dr. Catherine Hogan at HoganC@cardiff.ac.uk