SHP2 Inhibitors for Treating Cancer
, by Suman Mukhopadhyay, Carmine Fedele and Benjamin G Neel

Suman Mukhopadhyay PhD, Carmine Fedele PhD, and Benjamin G. Neel MD PhD
The research was performed at the Laura and Isaac Perlmutter Cancer Center, NYU Langone Health, New York, where Benjamin G Neel is the Director. Carmine Fedele is now at the Oncology Department of Novartis Institutes for BioMedical Research, Cambridge, Massachusetts, and Suman Mukhopadhyay is a postdoc in the Perlmutter Cancer Center, New York University Langone Health, New York.
Decades of research have identified mutations that constitutively activate proto-oncogenes in various types of cancer. KRAS (and to a lesser extent, other RAS isoforms) is among the most frequently mutated driver oncogenes and is often associated with therapeutic resistance and poor prognosis [1-3]. KRAS mutations are seen in nearly 20% of all patients presenting to major cancer centers, including >90%, 50%, and 25–30% of pancreas, colon, and non-small cell lung cancers, respectively [4, 5]. Mutant KRAS activates a complex array of pathways, in which cross-talk, feedback loops, branch points, and multi-component signaling complexes are recurrent features [1, 6]. Although several attempts been made to use potent and selective inhibitors of the ERK MAPK cascade (RAF/MEK/ERK) to target RAS-mutant malignancies, the efficacy of these small molecules is often negated by activation of upstream signaling mediated by these feedback loops, which results in ERK reactivation [7]. On the other hand, targeting parallel signaling pathways (PI3K/mTOR + MEK) has been limited by toxicity [8].
Earlier studies showed that MEK-inhibitor (MEKi) treatment of KRAS-mutant cancers results in “adaptive resistance”, characterized by induction of genes encoding receptor tyrosine kinases (RTKs) and/or their ligands [9-11]. Distinct RTK ligands are induced in different tumors, even within a single histotype, making it difficult to improve outcomes by combining RTK and MEK inhibitors. SHP2, a ubiquitously expressed non-receptor protein-tyrosine phosphatase encoded by the PTPN11 gene, lies downstream of almost all RTKs and is required for RTK-evoked RAS activation [12]. These features suggested that SHP2 inhibitors (SHP2i) might be attractive candidates for combination therapy to enhance MEK inhibitor (MEKi) action in KRAS-driven cancers. Subsequently, multiple groups, including ours [13-18], provided pre-clinical data supporting the idea of tackling “adaptive resistance” to MEKi by using SHP2i. Several pharmaceutical companies have developed SHP2 inhibitors, with multiple agents, including TNO155, RMC-4630, JAB-3068, RLY-1971, ERAS-601, and BBP-398, currently in phase 1/2 trials. One of these, RMC-4630, has been combined with MEKi in patients, and responses have been reported (7 patients treated, 1 stable disease, 1 partial response), but this combination might ultimately be limited by toxicity [19].
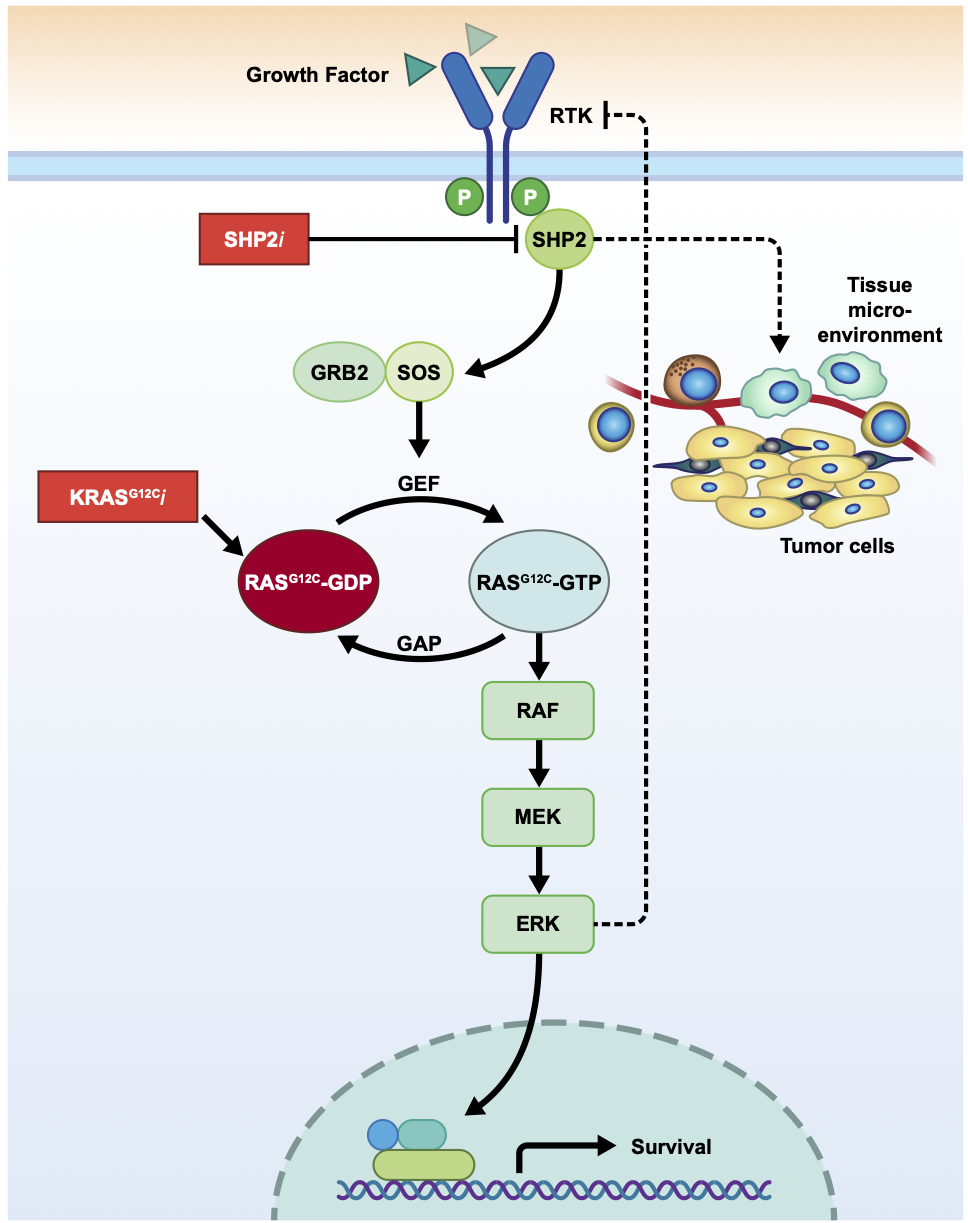
KRASG12C mutant with SHP2 + KRASG12C inhibitors
Pairing SHP2i with a mutant-selective agent might be a more promising therapeutic strategy, as it would limit the toxicity of “vertical” pathway inhibition seen with MEKi/SHP2i combinations. Previous studies showed that after MEKi-treatment of KRAS-mutant cancer cells, SHP2is prevent MEK/ERK pathway reactivation at the level of SOS1/2 [13, 18]. KRAS mutants differentially affect the RAS-GTP/GDP cycle, with some (most notably, KRASG12C) retaining significant intrinsic GTPase activity, although they remain defective in, or impervious to, RAS-GAP action [20, 21]. The residual GTPase activity in such “cycling mutants'' renders them dependent on SOS1/2 activity (exchange of GDP for GTP). Accordingly, in the MEKi/SHP2i combination studies, SHP2i efficacy correlated with the level of residual GTPase activity [13, 18], with SHP2is showing significant single agent activity against KRASG12C-mutant cancer cells [18, 22].
Long considered “undruggable”, the pioneering work of Kevan Shokat opened the door to the development of mutant KRAS-specific inhibitors, particularly targeting KRASG12C [23]. These landmark developments have led to several clinical grade KRASG12C inhibitors, the most advanced of which (AMG510, MRTX849) have shown significant single agent efficacy, particularly in NSCLC patients [24-27]. Lessons learned from studies of other targeted agents [9, 28, 29], including other inhibitors in the RAS/ERK pathway, suggested that drug resistance will also limit the effects of KRASG12C inhibitors (hereafter, G12Cis). This prediction has now been borne out in multiple pre-clinical studies [30-35], as well as in the clinical data reported on AMG510 and MRTX849 [36, 37]. Delineating G12Ci resistance mechanisms is essential for developing ways to prevent or combat their emergence.
Recent studies by others [25, 31, 32, 35, 38], along with our recent report [22], reveal that KRAS-mutant cancer cells develop adaptive resistance to G12C inhibitors (G12Ci). As is the case following exposure to other RAS/ERK pathway inhibitors (BRAFis/MEKis), genes encoding several RTKs/RTK ligands are induced upon G12Ci treatment. The addition of SHP2is improves G12Ci efficacy in several ways. First, G12Cis target the GDP-bound state of KRAS (KRAS-GDP). Effectively, then, G12Ci action is opposed by RAS-GEF activity. By preventing SOS1/2 action, SHP2is increase the amount of KRAS-GDP and thus the “target” of G12Cis [13, 22]. SHP2is also inhibit reactivation of other, wild type RAS isoforms (encoded by the other, normal, KRAS allele and/or H/NRAS) [31]. Consistent with these actions, SOS inhibitors also potentiate G12Ci action. For example, BAY-293, a potent SOS1 inhibitor, synergizes with ARS-853 to inhibit RAS activation and cell proliferation [39].
Most of the above studies on RAS/ERK pathway combinations either used standard cancer cell lines, cell-derived xenografts (CDXs), or patient-derived xenografts (PDXs). Such models, of course, lack a functional immune system and thus a bona fide tumor microenvironment (TME). Cancer cells and cells in the TME engage in multiple, complex, multi-dimensional interactions that affect tumor behavior and drug response [40, 41]. In our view, curative cancer therapies almost certainly require the generation of durable anti-tumor immune response. Viewed through this lens, the inability of any current targeted therapy/therapeutic combination to achieve cure reflects the failure of these strategies to generate such a response. Consequently, understanding the effects of any new agent/combination on tumor cells and the TME is essential if we are to move from responses to cures. This issue is particularly important for understanding the effects of inhibitors of targets that, like SHP2, act on immune receptor and cytokine signaling as well as RTK pathways in multiple TME cell types [42].
To this end, we investigated the cell-autonomous and non-autonomous effects of SHP2is alone and combined with G12Cis in syngeneic, genetically engineered mouse models (GEMMs) of pancreatic ductal adenocarcinoma (PDAC) and non-small cell lung cancer (NSCLC) [22]. As expected, each agent/combination had some similar effects in the two models. In both, the G12Ci/SHP2i combination elicited apparently favorable effects on the immune TME, decreasing myeloid suppressor cells, increasing total and CD8+ T cells and in most, also increasing intratumor B cells. In both models, intratumor T cells showed evidence of “exhaustion”, and anti-tumor efficacy in the PDAC model could be enhanced by addition of anti-PD1 antibodies to the G12Ci/SHP2i combination.
Other responses were quite different between these models. For example, as we reported in our earlier studies of MEKi/SHP2i combinations in PDAC [18], SHP2i alone had anti-angiogenic and anti-fibrotic effects in the PDAC model. By contrast, angiogenesis was increased in lung tumors from mice treated with single agent SHP2i or G12Ci/SHP2i combinations. The two NSCLC models, KRASG12C (KC) and KrasG12C;Tp53R270H (KCP), also differed in overall response and detailed effects on the TME.
To begin to deconvolve the locus of SHP2i action in the PDAC model, we generated tumors from cells reconstituted with a drug-resistant PTPN11 mutant (PTPN11T253M/Q257L). Tumors from cells expressing this mutant failed to regress or show influx of CD8+ or CD4+ lymphocytes upon SHP2i treatment. This lack of anti-tumor immune response correlated with (and was likely caused by) failure to alter the expression of genes encoding specific cytokines and chemokines. Despite the lack of significant regression, however, there was a marked “treatment effect”, characterized by a reduction in tumor cell number and collagen scarring. We attributed these findings to necrosis induced by the direct anti-angiogenic effects of SHP2i. By contrast, the anti-fibrotic effects of SHP2i were reversed in tumors expressing the drug-resistant SHP2.
These data illustrate that tumor genotype (including mutational burden), cell-of-origin/histotype, and intrinsic and extrinsic features of the TME play key roles in the response to targeted therapies, and argue that rational combination therapies must consider all of these effects. Notably, our results differ in detail from other reports [26, 43, 44] that evaluated syngeneic tumor models (e.g., CT26) injected sub-cutaneously, unlike our orthotopic or autochthonous models. Studies combining AMG510 or MTRX849 with targeted or immune therapies, including SHP2is (NCT04330664, NCT04699188, NCT04185883), MEKis (NCT04185883), or anti-PD1(NCT04613596, NCT03785249, NCT04185883) are underway, as is a trial of MRTX849 in combination with the SOS1/pan-KRAS inhibitor BI 1701963 (announced by Boehringer Ingelheim, December 2020). It will be interesting—and important—to determine how these combinations affect the TME as well as the tumor cells themselves, and how well our mouse models predict the human response. Finally, with inhibitors of other mutant KRAS alleles already announced (MRTX1133-KRAS G12D selective inhibitor, Mirati Therapeutics Inc., October 2020 ) or in active development (KRAS-G12D(ON) Inhibitors, Revolution Medicines Inc., September 2020), it will be critical to determine whether the specific mutation also influences the TME response and thus the combination(s) most likely to be optimal for patients.
For further information, contact Dr. Benjamin Neel at benjamin.neel@nyumc.org.