RAS-binding compounds: approaching the undruggable from a different perspective
, by Terry Rabbitts

Terry Rabbitts, PhD,FRS, FMedSci
Terry Rabbitts is Professor of Molecular Immunology in the Centre for Cancer Drug Discovery, Institute of Cancer Research, Sutton, London, UK.
There are numerous examples in biomedical research of progress toward apparently unattainable objectives through the development of new approaches. One such biological target was RAS, where decades of attempts had failed (until very recently) to yield anti-cancer drugs directly interfering with RAS function. We adopted a distinct approach aimed initially at interfering with protein-protein interactions of RAS, and other so-called undruggable targets like transcription factors, using molecular biology tools called intracellular antibodies that bind to target proteins inside cells. These tools were initially used in RAS target validation and subsequently for isolating drug leads, guided by the intracellular antibody interaction with RAS.
Our work on intracellular antibody fragments as tools for analysis of protein complexes argued that these macromolecules could also be employed for target validation in diseases and then as guides for target-specific surrogate compounds as inhibitors. The former issue was often neglected prior to initiating drug discovery programmes while the latter was considered impossible because it was purported that antibodies were unsuitable to derive compounds. We wondered if these difficulties could be overcome by focussing on the antibody combining site (i.e. the part of antibody interacting with antigen) to guide the derivation of chemical compounds that would be surrogates of the antibody, because the combining site is the part relevant to the protein-protein interaction between antibody and antigen. It had been thought however that antibodies were too large for this purpose. Nonetheless, early work from the laboratory of Cyrus Chothia and others [1-3] showed that the interface areas of antibody-antigen interactions have a mean of about 1600 Å2. This seemed much larger than would be expected for the range of space occupied by drug-like chemical compounds, estimated at 300 to 1000 Å2 [4, 5]. However, small intracellular antibody fragments such as single variable Fragments (scFv, comprising antibody heavy chain and light chain variable regions) or intracellular domain antibodies (iDAbs, comprising only a single variable region) reduce the interface area. Based on these factors, it seemed possible that compounds that bind to antigens could be selected using Dab binding sites and that these compounds would be surrogates of the Dab. The corollary is that the specific interaction interface between the antibody fragment with target protein would be mimicked by the chemical compound surrogates.
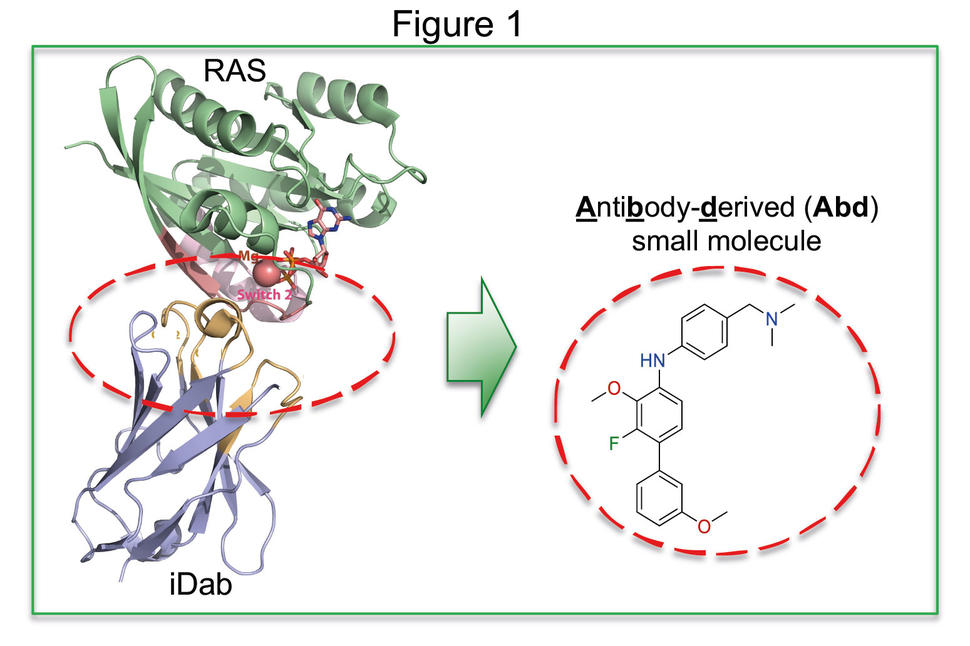
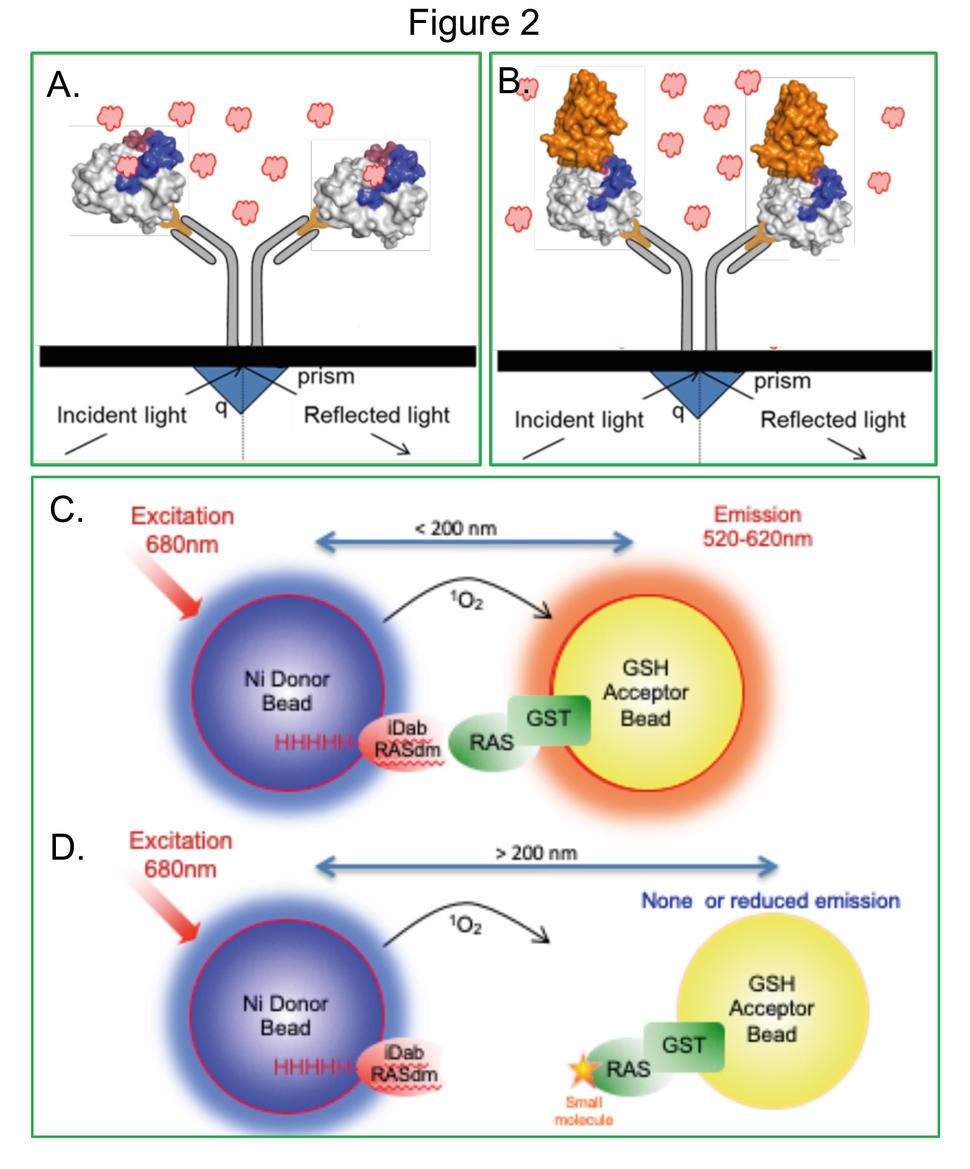
We developed this concept by applying an intracellular antibody fragment that binds to activated RAS isoforms [6], interferes with RAS-effector protein-protein interaction and signal transduction [7], and inhibits growth of RAS mutant cells in pre-clinical models [8]. We exploited the specific binding of the antibody fragment and were able to identify RAS-binding compounds in antibody-competition assays (illustrated in Fig.1). The method was called Antibody-derived (Abd) technology. In the first Abd technology application, we used a competitive surface plasmon resonance (cSPR) method [9] that depends on high affinity interaction of the antibody fragment with GTP-bound RAS. When the antibody fragment is bound to RAS, it serves as a competitor of compounds binding to the same region. This competitive method is illustrated in Fig. 2A-B (panel A depicts a compound binding to RAS; panel B depicts inhibition of this compound by the presence of the anti-RAS antibody fragment). Thus, our Abd technology identified a hit chemical series interacting with a pocket [10, 11] near the effector binding region in RAS (activated pan-RAS binding compounds) [9]. Using X-ray crystallography soaking of KRASQ61H crystals by compounds, we implemented a structure-based compound development programme [9, 12]. This work demonstrated two points. First that RAS is indeed druggable (as many labs have now shown, in particular KRASG12C binding compounds [13-15]) and that antibodies could indeed be used to guide the isolation of chemical matter with our Abd technology. The latter discovery was important for us because it suggests that the antibody combining sites could generally be accessible for implementing the Abd technology approach to drug discovery.
The chemical compound series that was developed in this work is a pan-RAS series binding to all the RAS isoforms [12] because the binding pocket is found in KRAS, HRAS and NRAS. This followed from our intracellular antibody being pan-RAS binding, although with substantially higher binding to activated (GTP-bound) RAS than GDP-RAS in cell-based assays [7]. The therapeutic index of such compounds, if developed into drugs, would depend on several factors such as the susceptibility of cancer cells expressing mutant RAS compared to normal cells that may be quiescent in vivo or undergoing cell division such as in the gut epithelium. An exciting development in the intracellular antibody technology is RAS-isoform specific intracellular antibodies that we have developed following a study of KRAS-specific Designer Ankyrin repeat proteins (DARPins) [16]. We have recently isolated KRAS-restricted scFv that require the presence the KRAS-specific residue histidine 95 for binding. This means that it is feasible to select residue-selective anti-RAS intracellular antibodies that should constitute a tool-box of isoform specific reagents for use in Abd technology, and perhaps also potentially mutant-selective ones.
On the whole, antibodies are most efficacious in high affinity form, where the Kon is high and Koff is low, so that their binding to a target is sustained in time. This property can be an advantage in bioassays of protein function and also in Abd competition screening, as we did with RAS [9]. A disadvantage is that the low dissociation constant Kd value makes it difficult to directly identify compounds in a chemical library of sufficient affinity to interfere with the interaction of antibody and target. Accordingly, in a further development to Abd technology, we employed an approach in which the binding affinity of the antibody was reduced (dematuration) to a level that allows RAS-binding compounds to directly interfere with the interaction of antibody and RAS [17]. Thus this application of Abd technology relies on direct inhibition by compounds of antibody binding to antigen, unlike the cSPR approach [9].
An Abd screen application was achieved employing the dematured version of the anti-RAS scFv [17]. This intracellular antibody-RAS screen was implemented in an AlphaScreen assay (depicted in Fig. 2C, D). Interaction of dematured antibody with RAS generates a signal (Fig. 2C). Competition of this interaction by chemical compounds was directly assessed with a chemical library where compounds that bind to RAS interfere with antibody-binding, reducing the Alpha signal (Fig. 2D). In this proof-of-concept application of Abd technology, we identified two RAS-binding compounds from two commercial libraries (2560 compounds) [17]. The general significance of implementing this version of the Abd approach is that target validation can first be achieved using a high affinity intracellular antibody prior to dematuration for use as a tool for drug discovery. This is one reason why employing flexible reagents such as antibody fragments is advantageous over using natural protein-protein interaction partners. In addition, no structural data are needed for antibody dematuration since the interaction zones of variable regions are largely defined by the complementarity determining regions in the antibody variable region primary sequences.
The prospective applications of Abd technology extend beyond the mutant RAS problem in cancer because using high affinity intracellular antibodies can be employed in functional studies and disease target validation, prior to and followed by drug discovery. This gives a landscape of opportunities to combine molecular biology/molecular immunology with chemical biology for drug discovery in the range of human cancers but also in other human diseases.