New Clarity on the Warburg Effect
, by Alba Luengo, Zhaoqi Li, and Matthew Vander Heiden

Alba Luengo PhD, Zhaoqi Li PhD, and Matt Vander Heiden PhD.
This research was performed at the Koch Institute for Integrative Cancer Research at MIT, where Matt Vander Heiden is Associate Director. Alba Luengo is now at Toran Therapeutics of the Flagship Pioneering Group, and Zhaoqi Li is a postdoc in the Department of Microbiology, Harvard Medical School.
Nearly a century ago, Otto Warburg discovered that tumors consume tremendous amounts of glucose relative to most non-transformed tissues, and that the majority of glucose consumed by tumors is fermented to lactate, rather than oxidized in pathways that require respiration [1]. This phenotype is referred to as “aerobic glycolysis,” because unlike carbohydrate fermentation in response to oxygen limitation, aerobic glycolysis involves high levels of fermentation even when oxygen is abundant. Aerobic glycolysis a hallmark of proliferative metabolism found across many kingdoms of life, but is frequently associated with cancer cells, and is known as the Warburg effect in this context.
-Luengo, et al., 2020 Mol Cell Dec 22.
What drives aerobic glycolysis, and why it is associated with proliferation, has been a long-standing question [2,3]. Proliferating cells require ATP to meet increased demand for biomass synthesis, and yet aerobic glycolysis is less efficient for ATP production than oxidative metabolism. One proposed explanation for the phenotype is that increased glucose consumption provides important biosynthetic precursors for anabolic reactions branching from glycolysis, including pathways that produce lipids, nucleosides, or proteins. However, the majority of glucose carbons consumed by cells are excreted as lactate. Indeed, the majority of the biomass of proliferating cells is derived from amino acids, rather than glucose [4]. Another explanation that has been put forth is that aerobic glycolysis facilitates production of electron carriers required as cofactors for redox reaction in cells. Increased glucose uptake permits higher production of the reducing equivalent NADPH by the oxidative branch of the pentose phosphate pathway, and fermentation involves regeneration of the oxidizing equivalent NAD+ via the action of lactate dehydrogenase (LDH). Other models have suggested that aerobic glycolysis optimizes for ATP production [5,6], or results from molecular crowding [7]. A definitive explanation for why aerobic glycolysis is associated with cell growth has been lacking and is an area of active investigation.
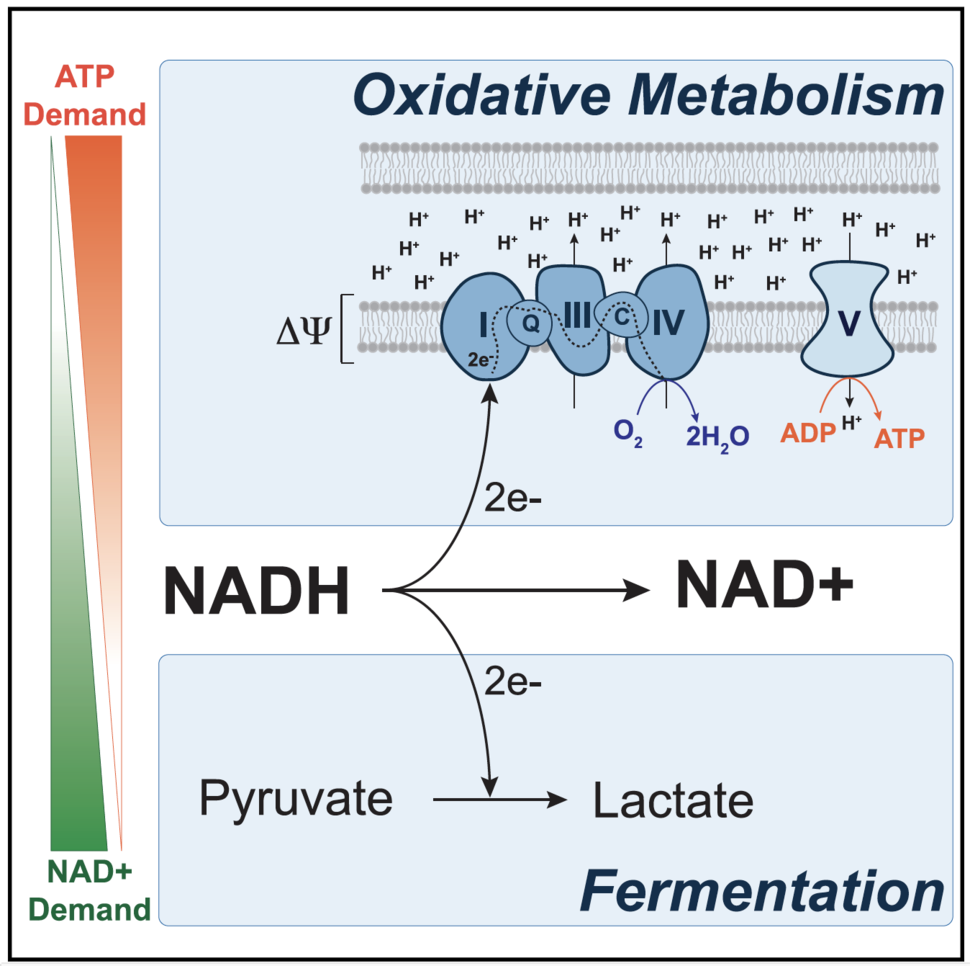
The metabolic fate of pyruvate determines the extent to which cells engage in aerobic glycolysis. Pyruvate is the end-product of glycolysis and lies at the intersection of glycolysis in the cytosol and the tricarboxylic acid (TCA) cycle in the mitochondria. Pyruvate oxidation requires the pyruvate dehydrogenase complex (PDH), which facilitates the entry of pyruvate carbons into the TCA cycle. PDH is negatively regulated by the pyruvate dehydrogenase kinases [PDK], and PDK inhibition suppresses aerobic glycolysis by promoting oxidation of glucose carbons in the TCA cycle at the expense of fermentation. Genetic suppression of PDKs has been shown to slow cancer cell growth in culture and in tumors [8,9], and we used PDK inhibition to study the metabolic underpinnings of how aerobic glycolysis supports proliferation [10].
We determined that suppressing fermentation by PDK inhibition decreased capacity of cells to produce the cofactor NAD+, a product of the LDH reaction. NAD+ is a critical electron acceptor required in many cellular processes and to synthesize oxidized biomass molecules, including lipids, nucleotides, and some amino acids, including aspartate [11-13]. We found that endowing cells with various means to regenerate NAD+ independently of LDH restored proliferation rates in cells in which aerobic glycolysis was suppressed, confirming that fermentation supports proliferation by promoting NAD+ regeneration. However, these results were surprising since the mitochondrial electron transport chain (ETC) can regenerate NAD+ when oxygen is available. Given that our experiments were performed in normoxia, we faced an unanticipated question: why is the ETC inadequate to maintain NAD+ homeostasis in some rapidly proliferating cells?
The ETC does not function in isolation. It is part of a series of reactions collectively known as oxidative phosphorylation, whereby the transport of electrons through the ETC generates a proton gradient across the inner mitochondrial membrane. We wondered whether PDK inhibition could cause the proton gradient to become too large, causing ETC to be thermodynamically “product inhibited.” We found that dissipating the proton gradient increased ETC activity, NAD+ regeneration and proliferation, suggesting that ETC was indeed constrained by the proton gradient when aerobic glycolysis was suppressed.
In eukaryotic cells, the potential energy stored in the mitochondrial proton gradient is harnessed by ATP synthase to phosphorylate ADP to make ATP. Our finding that an elevated proton gradient can function as an inhibitor of the ETC led us to hypothesize that the endogenous mechanism of proton gradient dissipation, ATP synthase, was somehow inadequate in its capacity to relieve this thermodynamic block on the ETC. One way to increase the rate of proton flow through ATP synthase is to increase ADP levels. To do so, we treated cells with a sublethal dose of the ionophore gramicidin, which facilitates the flow of Na+ and K+ ions through the plasma membrane and elicits the cell to engage in elevated Na+/K+ ATPase activity to maintain the Na+/K+ gradient at the plasma membrane. The net effect of this elevated activity is an increase in ATP hydrolysis in the cell, which supplies ADP for ATP synthase to use as a substrate. We found that treating cells with gramicidin relieved the elevated mitochondrial proton gradient, which ultimately allowed NAD+ regeneration by ETC and proliferation with decreased dependency on fermentation. These experiments led us to conclude that because oxidative phosphorylation is coupled and generates NAD+ and ATP at a fixed stoichiometry, a failure to hydrolyze ATP limits ATP synthase activity and constrains NAD+ production by the mitochondrial ETC.
Aerobic glycolysis is an inefficient means of generating ATP when considering yield per glucose molecule. However, our data suggests that ATP is not necessary limiting for proliferation, and that excess ATP can instead impose an upper limit on the oxidative capacity of mitochondria. Several perplexing studies have found evidence that futile metabolic cycles can promote cell proliferation. For example, cancer cells that do not express the tumor suppressor PTEN upregulate enzymes promoting ATP wasting [14], and elevated ATP has been shown to impair tumor growth [15]. The finding that insufficient ATP synthase activity can restrict NAD+ regeneration could potentially explain why pathways that promote ATP consumption could also promote proliferation.
While fermentation allows for NAD+ regeneration uncoupled from ATP synthase, glycolysis is redox neutral and does not net regenerate NAD+. Thus, why increased glucose uptake and glycolysis are also often associated with proliferation remains an open question. Nevertheless, our model to explain why many proliferating cells engage in aerobic glycolysis is consistent with other observations in cancer. Aerobic glycolysis is best characterized by a shift of pyruvate carbons away from the TCA cycle and towards fermentation, rather than an upregulation of glycolysis at the expense of oxidative phosphorylation. This distinction is a subtle but important one, as ETC activity has been showed to be required for tumorigenesis and cell proliferation [16, 17]. Proliferating cells have high requirements for NAD+, as this cofactor is needed to catabolize reduced nutrients and to synthesize oxidized biomolecules. Our study shows that in regimes of high NAD+ demand, ATP synthase activity constrains NAD+ regeneration by the ETC and fermentation, rather than pyruvate oxidation, is preferred as a supplementary NAD+ producing pathway [10]. Therefore, aerobic glycolysis reflects a metabolic state in which the demand for NAD+ for oxidation reaction supersedes the cellular demand for ATP.