The use of Molecular Docking as a ligand discovery tool; Can machine learning help the pursuit for ligands?
, by Trent E. Balius and Megan Rigby

Trent E. Balius is one of the developers of the UCSF DOCK software, which is a computational tool used to predict how a small molecule (ligand) binds a site on a protein (or other macromolecule). Trent leads the RAS Computational Chemistry team at the NCI RAS Initiative, where he uses molecular docking to search for novel RAS-targeted therapeutics. He received his PhD from Stony Brook University studying Computational Biology in the Department of Applied Mathematics and Statistics under the direction of Robert C Rizzo, and performed his postdoctoral training in the Shoichet Laboratory, in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
Megan Rigby is a member of the Covalent Inhibitors group at the RAS Initiative as a Research Associate II. She is currently completing her graduate studies in Biomedical Science at Hood College.
Interview (Edited Transcript).
Megan: Thank you for agreeing to share your work with our readers, Trent! Let’s dive right in—how do you decide what molecules to focus on in computational ligand/drug design and discovery? What small molecules do you screen?
Trent: I believe in focusing on molecules that are accessible—molecules that are available to buy and are inexpensive. I have this philosophy ingrained in me by my training in the Shoichet Lab at UCSF that computational methods are only useful if you can TEST the prediction.
I perform a large docking screen where I dock hundreds of millions of small molecules into a protein pocket and then our group will predict whether some will bind, and then we test it experimentally. So, if you want to do that experimental testing, which is the only way a virtual screen is useful, then you need to be able to buy the molecules (in stock) or make the molecules (make on demand). With in-stock molecules, they are sitting on a shelf somewhere, and you can request a quote for those compounds. Make-on-demand molecules do not exist yet, but the company has building blocks/pieces of molecules that they will react together to form a product, and then they’ll sell you that product. What’s most important for accessibility is that the molecules are inexpensive and can be made.
Megan: Do you see in silico screening being a big player in the search for RAS therapeutics, and what has been its role so far?
Trent: I think computational methods can play a role in targeting RAS. In a lot of the projects that are going on in the RAS Initiative now, like in our partnerships with drug companies and other national labs, there’s a lot of computational work happening, including docking and molecular dynamics simulations. To focus on one project, I’m working with Simanshu and his team. They have some crystal structures of a specific mutant of RAS with fragment molecules bound, and I’ve done virtual screening against those crystal structures. We are exploring SAR (structure-activity relationships) around those docking hits as well as with the original fragments, and found more binders through buying analogs of those virtual screening hits (or making them by working with David Turner and Team).
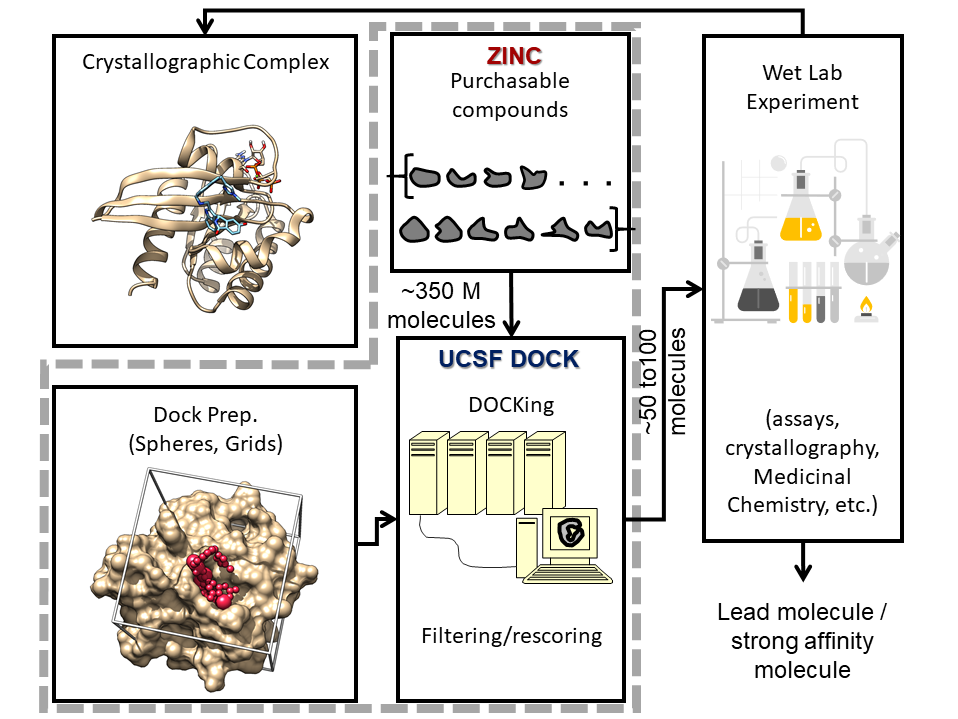
UCSF DOCK software is used to screen millions of purchasable compounds by predicting their ability to bind to active RAS. Hits are then evaluated at the bench using in vitro or cellular assays, crystallography, and other techniques.
Megan: What are some of the challenges you’ve faced with RAS, in particular, as opposed to other proteins?
Trent: First of all, RAS is a flexible protein. Also, RAS has a cofactor, it has either GTP or GDP. If it has GTP, it’s almost always an analog in the crystal structures (a non-hydrolyzing analog).
Megan: Right, because the real target is GTP-bound RAS—the oncogenic state.
Trent: Right, the GDP (in the off state) or GTP (in the on-state) influence the conformational state of RAS as do cancer associated mutations which usually lock RAS in the on-state.
So, the cofactor is a layer of bookkeeping that I have to pay attention to when preparing the protein for docking. I have to assign partial charges to the cofactor, and I now have automated procedures to prepare the cofactor. So when I do the parameterization, I also have to make sure that the protonation states are correct, that the hydrogens are on the right atoms, and that the partial charges are correct. It seems like there’s a lot of conformational change that can happen within RAS, and typically in docking we restrict ourselves to a single state for the screen, so that can be an issue. So, two things that make modeling RAS difficult are preparing the cofactor, and RAS flexibility.
Megan: What helps you decide one crystal structure rather than another to run your screen on?
Trent: Often, crystal structures that are already bound to a ligand perform better in docking screens. So right now, a lot of the screens that I’ve done are from in-house crystal structures from Simanshu’s team.
Megan: Screening in silico first seems like it would significantly raise the success rate—how effective do you find it?
Trent: The interesting thing about molecular docking as a drug discovery tool is that docking is not very accurate. If you buy 100 molecules that dock well and were selected from a list of “hits” by an expert, at most you’ll get 30 molecules that will bind, more likely you’ll get 15 molecules that will bind.
If you were designing something like a car or an airplane using engineering principles, then our methods are lousy in comparison. If somebody’s coming to me and asking me to make one prediction, and then they spend thousands of dollars making my one prediction and it doesn’t work, that person is going to be upset. Since this is the case with docking, we need multiple shots on goal. So, we would expect somewhere between a 2-30% success rate for a set of molecules selected from a docking screen and tested experimentally.
Megan: From a traditional high throughput screening perspective, though, that’s a pretty high success rate…
Trent: But it is not as good as we would like. We’re only testing a fraction of what we screen. So docking is this filter. You’re sifting through many molecules, and then you take the top of the list to visually inspect. The final step is the human filter to select the molecules.
Megan: How long does it usually take to come up with an initial library? How many compounds do you screen at a time?
Trent: I use the ZINC database from the Shoichet and Irwin labs at UCSF. John Irwin and Brian Shoichet created the ZINC database in the mid 2000s. ZINC stands for ‘ZINC is not commercial,’ and consist of molecules collected from vender catalogs. There is a webpage for the ZINC database where you can download molecules that are prepared for computational analysis. ZINC22 will be a revamp, and it will have a lot more molecules. Accessible chemical space has been growing tremendously because of these make on demand catalogs and so too has ZINC. This is a good problem to have, but is a little intimidating. The questions are how big is too big? And how can we keep up?
For reference, if I dock lead-like compounds (a certain slice of chemical space with a molecular weight and hydrophobicity range), which is about 330 million molecules with my copy of ZINC[1][2], I spend about 1 second per molecule, and I’m using about 1,000 CPU cores. Then, it takes about 3 and a half to four days. Now, let’s scale up. If I dock half a billion, then that’s 5 days, and if I dock a billion, then that’s 10 days. If I dock ten billion, that’s 100 days. We can’t afford to spend that much time on one screen. The only way that would be feasible is if we made the software faster so we can get the results in a more reasonable time.
Megan: So, will there be a limit to the number of compounds in one screen, or are there things people are doing to make the process more efficient?
Trent: One thing that other people are doing is using machine learning to train on a smaller set of molecules, so they’ll do a screen and then they’ll train a machine learning model on that smaller screen, and then they’ll extrapolate that to a larger screen, and then they’ll dock the ones that score well. So they create a pseudo-docking method[1][2]. I also saw a talk where they did a sparse screen, and then they focus in on different sections of the database, so they’ll do a dynamic screen where they screen a random set and then based on that, they’ll zoom in on a region to screen, so they won’t screen the whole database, they’ll focus in on the portion that looks interesting.
Megan: Do you see AI driving improvements in the docking process moving forward?
Trent: Yes, I think there are a lot of believers in AI. The challenge with machine learning and AI is you need a lot of data to train it on. And often in drug discovery, the data is sparse.
Drug discovery is hard for a number of reasons. Just binding is difficult to understand, but then you have all these other confounding factors, like aggregation, which is when small molecules aggregate together and lead to issues. And you also have these weird things, like ‘affinity cliffs’ where a small change in the ligand chemical structure can abolish binding. Another problem is that the scientific literature is riddled with artefacts. So, people will claim that these molecules bind, and they don’t actually bind the way people think they do. For instance, many molecules bind non-specifically, and they are having a cellular effect, but they are not acting through the mechanism that people believe.
So, for machine learning to be an effective tool for ligand discovery, you need clean data and you need lots of data. The other concern about deep learning—neural networks—is you don’t know what’s happening under the hood.
Machine learning may make things better, but I don’t think it’s going to solve all our problems.
Megan: Right, it does seem like there’s a lot of unmeasured excitement around AI right now. So, to be clear, if two people trained a machine learning algorithm on two different datasets, they could come up with two completely different results?
Trent: Yes, the limitations of machine learning is that your training set is going to control what you’re going to find. So let me tell you my bias: I’m very much in favor of physics-based models. I think these AI machine learning methods are really useful and helpful, but they’re knowledge-based, they’re not physics based, and as you say, it’s all about the data that you include. However, there are people that are also using AI to train physical models to get the physics right.
I think that this idea of using different scoring functions for different proteins is unsatisfying. It’s much more satisfying to think that we’re getting the physics right. Don’t get me wrong, though, machine learning is very useful.
Megan: I can also see what you mean about the limitations of knowledge-based approaches—like with protein structure, there’s still a lot of unknowns regarding conformational states across RAS mutants and GDP/GTP states…
Trent: Yes, so I’ll just give you an example of a machine learning method that has changed the field which is alpha fold. Alpha fold is responsible for a leap in our ability to predict protein structure. In fact, some people are saying the protein folding problem has been solved. You can give alpha fold a sequence and it will predict a structure. But as you just said, for RAS, it’s a very flexible protein. There are the loop states that change conformation. There’s a bias towards different states—whether it’s GDP and GTP, the conformation will look different. And then you have effector binding and things like that. Although alpha fold can predict a protein fold, it doesn’t show what’s binding to it. Interestingly, it leaves the nucleotide binding site there without nucleotide. I’ve been using alpha fold for modeling proteins and protein complexes and it’s a HUGE advance.
Megan: Do you have any final thoughts?
Trent: The future is bright for computational methods aiding drug discovery, with improvements happening from different directions. Growth in chemical space, advancement in methods (including AI), improvements in hardware (like more diskspace, and GPUs). Yes, I think the future is bright for computational drug (ligand) discovery.