KRASG12C inhibition drives anti-tumour immunity in lung cancer but combinations with anti-PD1 immunotherapy may only benefit patients with ‘inflamed’ tumours
, by Jesse Boumelha, Edurne Mugarza, Sophie de Carné Trécesson, Febe van Maldegem, Miriam Molina and Julian Downward Francis Crick Institute, London
Jesse Boumelha works in Julian Downward’s lab at the Francis Crick Institute, where he was recently awarded his PhD. His research focuses on understanding mechanisms of immune evasion in novel mouse models of KRAS-mutant lung cancer in order to develop rational immunotherapy combinations.
Edurne Mugarza is a former graduate student in the Downward lab at the Francis Crick Institute working on KRAS-G12C inhibitors and their influence on the tumour microenvironment and anti-tumour immunity. She is currently a postdoctoral researcher at UCLA with Cristina Puig Saus.
Sophie de Carné Trécesson is a postdoc in the Downward lab at the Francis Crick Institute where her work focuses on how KRAS signaling shapes the tumour microenvironment using single cell technologies.
Febe van Maldegem was a postdoc in the Downward at the Francis Crick Institute until last year and now leads her own lab in the Department of Molecular Cell Biology & Immunology at Amsterdam University Medical Centre. She works on investigating the interactions between the tumour and immune system in lung cancer, with an emphasis on the spatial organisation of the tumour microenvironment.
Miriam Molina-Arcas is a principal laboratory research scientist in the Downward laboratory at the Francis Crick Institute. Her research is on the identification of therapeutic strategies for the treatment of lung cancer tumours harbouring KRAS mutations.
Julian Downward’s research has focused on RAS oncogenes for many years, first at Cancer Research UK and for the past six years at the Francis Crick Institute in London. His ambition is to fully understand the interplay between RAS mutant cancers and the immune system and to use this knowledge to cure advanced cancers using combinations of KRAS inhibitory drugs and therapies targeting the immune system.

(left to right) Miriam Molina-Arcas, Jesse Boumelha, Julian Downward, Sophie de Carné Trécesson, Edurne Mugarza, and Febe van Maldegem
It has been 40 years since the seminal discovery of RAS oncogenes, which kickstarted a new era of cancer biology that defined tumourigenesis as a genetic disease involving the dysregulation of normal cellular processes. This led to a rapid advancement in our understanding of the molecular mechanisms underlying tumourigenesis, in large part through the use of genetically engineered mouse models (GEMMs), solidifying the causal role of oncogenic mutations as drivers of the malignant phenotype. The majority of these studies focused on the cell-autonomous contribution of oncogenes to cell proliferation, survival and metabolism. However, accumulating evidence suggests that oncogenic signalling can engage host cells within the tumour microenvironment (TME) and dampen immune responses which, given the clinical success of immunotherapy, have been recognised as having powerful anti-tumoural functions. However, the effects of oncogenic KRAS on the TME have been difficult to study due to the lack of suitable tools. Pioneering work led by Kevin Shokat over the past 10 years has resulted in the remarkable development of mutant-specific KRASG12C inhibitors which directly inhibit KRAS, hitherto considered ‘undruggable’, by binding to the thiol group of the novel cysteine residue, trapping it in an inactive state. Whilst such a drug had massive clinical implications, it also could be used as a preclinical tool to specifically inhibit oncogenic KRAS signalling in tumour cells to probe the influence of tumour-cell intrinsic oncogenic signalling on the TME.
Using multiple immune-competent mouse models of KRASG12C-mutant lung cancer, we investigated the effect of KRASG12C inhibition on the TME and anti-tumour immunity (1). This work demonstrated that oncogenic KRAS signalling promotes immunosuppression via multiple mechanisms, including driving the expression of myeloid cell chemoattractants, inhibiting antigen presentation and directly inhibiting tumour-intrinsic interferon (IFN) signalling which is critical for anti-tumour immunity. Importantly, KRASG12C inhibition reversed KRAS-mediated immunosuppression, remodelling the TME with reduced infiltration of monocytes and neutrophils accompanied by influx of T cells with improved cytotoxic functions. Furthermore, tumour cell death promoted by KRASG12C inhibition promoted antigen uptake and T cell chemoattractant secretion by dendritic cells.
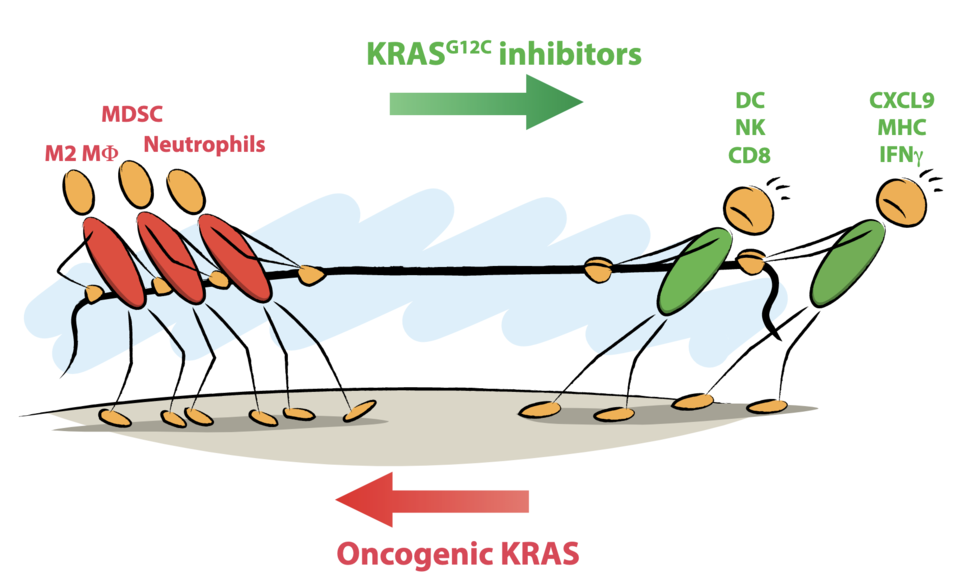
KRAS G12C-mediated immunosuppression is released by KRAS G12C inhibitors
The landmark development of KRASG12C inhibitors has been rapidly followed by the generation of clinical compounds, which have shown promising efficacy in early-stage clinical trials. Amgen’s sotorasib (AMG-510) was recently approved by the FDA for the treatment of locally advanced or metastatic KRASG12C NSCLC. Whilst sotorasib achieves impressive tumour control in a substantial proportion of patients, benefits are often short-lived with the rapid acquisition of resistance. Indeed, a recent phase III clinical trial comparing sotorasib to docetaxel demonstrated that whilst KRASG12C inhibition improved progression-free survival, it failed to extend the overall survival of patients. This is an issue shared with many other targeted therapies and is in stark contrast to the durable responses achieved in a subset of patients upon reinvigoration of anti-tumour immunity by immunotherapy. Therefore, a greater understanding on how KRAS inhibitors affect the TME and anti-tumour immunity will be crucial for developing rational therapeutic combination strategies.
Obtaining clinically meaningful insights requires the use of preclinical mouse models that reflect the range of immune landscapes seen in human lung cancer. The TME in human cancer can be classified into three main phenotypes: immune-desert (‘cold’), immune-excluded, and ‘inflamed’ (2). Inflamed tumours are infiltrated by numerous immune cells including cytotoxic T cells and are most likely to respond to immunotherapy whilst immune-excluded, where immune cells are constrained to the stroma, and ‘cold’ tumours, which contain few T cells, are normally resistant to immunotherapy. Preclinical mouse models of lung cancer are notoriously poorly immunogenic, displaying either ‘cold’ or immune-excluded TMEs and refractory to immunotherapy. Given that 20% of lung cancer patients are responsive to immunotherapy, we generated a novel immunogenic mouse model of KRAS-mutant lung cancer (3) by establishing cell lines from autochthonous KP tumours expressing the human DNA cytosine deaminase APOBEC3B to increase the mutational burden which is notoriously low in GEMM tumours. The resultant orthotopic model stimulated spontaneous anti-tumour immune responses and was partially sensitive to immunotherapy, although immune responses were, surprisingly, targeting endogenous retroviral antigens rather than APOBEC3B-induced neoantigens. This model has proven very useful for the study of processes that regulate anti-tumour immunity and the evaluation of targeted and immunotherapy combinations.
Using mouse models with different immune landscapes, we showed that KRASG12C inhibition reversed immunosuppression, inducing a pro-inflammatory transcriptional program and stimulating adaptive immunity in ‘cold’, immune-excluded and ‘inflamed’ lung tumours. This prompted us to assess the potential synergy of KRASG12C inhibition and immunotherapy which has already shown superior efficacy in a preclinical model of colorectal cancer. However, this combination only proved beneficial in the ‘inflamed’ lung cancer model, with a substantial subset of mice achieving durable responses. Similarly, we showed that adaptive immunity contributed to durable responses in mice treated with KRASG12C inhibitors but, again, only in ‘inflamed’ tumours. These findings have stark clinical implications, as it suggests that combining KRASG12C inhibitors with immunotherapy may only benefit patients with ‘inflamed’ tumours. Most clinical trials testing this combination, with the exception of KRYSTAL-7, primarily include patients who have progressed on immunotherapy and therefore any potential benefits may not be realised. Additional combination strategies may be required in order to sensitise ‘cold’ tumour types to immunotherapy and future studies using preclinical models of lung cancer that cover the range of immune landscapes observed in humans will be essential for uncovering these.
Whilst these preclinical studies have shed light upon the potential utility of combining KRASG12C inhibitors with immunotherapy in a subset of lung cancer patients, there is a more immediate problem which needs to be overcome. Amgen’s early-phase clinical trial combining sotorasib with immunotherapy was hampered by high-grade liver toxicities in a substantial proportion of patients. The source of these toxicities is still unclear but may be related to the excessive dosing at which sotorasib is administered, initially considered tolerable due to the mutant-specific nature of the drug. A key question is whether preclinical models will be useful for studying mechanisms underlying toxicities that arise from these combination therapies. Indeed, immune-related adverse events which are common in patients treated with immunotherapy are rarely observed in preclinical mouse models. Beyond this, a greater understanding of how oncogenic KRAS promotes immunosuppression, especially given recent advancements in single-cell technologies, could identify novel immune-combination strategies that may be able to improve outcomes for KRAS-mutant lung cancer patients.