Top-Down Proteomics Reveals the KRAS Proteoform Landscape in Colorectal Cancer

Neil Kelleher, PhD, Lauren Adams, PhD candidate, and Caroline DeHart, PhD
Lauren Adams is currently a senior Ph.D. candidate in Neil Kelleher’s laboratory at Northwestern University. She combines the precision of top-down mass spectrometry with molecular biology techniques to identify, characterize, and probe the functionality of KRAS proteoforms in cancer. Neil Kelleher is a professor at Northwestern University who has devoted his career to developing top-down mass spectrometry techniques and applying them to better understand proteoform-level biology. Caroline DeHart leads the mass spectrometry team for the NCI RAS Initiative at the Frederick National Laboratory for Cancer Research in Frederick, MD. She employs targeted top-down proteomics to study RAS proteoforms in various cancer contexts and applies complementary proteomic methods to elucidate the roles of post-translational modifications in oncogenic signaling pathways.
Proteomics is a broad term that can generically be defined as the study of the proteome, or the landscape of proteins expressed within a given biological context. More specifically, proteomics can entail investigating aspects of the proteome such as protein expression levels, post-translational modifications (PTMs), cellular localization, and interacting partner networks. There are several experimental approaches which are frequently used to interrogate the proteome, including both immunoblotting- and mass spectrometry-based techniques.
Immunoblotting is an attractive approach in that it does not generally require complicated upstream sample preparation, utilizes small sample volumes, and delivers relatively rapid results. However, immunoblotting is reliant on the availability of a sensitive and specific antibody for the protein and/or PTM of interest. In the case of the RAS family of GTPases (KRAS4A, KRAS4B, HRAS, NRAS), there remain few reliable isoform- or mutant-specific antibodies1. Thus, it can be quite challenging to discern which RAS isoforms may comprise a band, let alone which mutations or PTMs may be present within a RAS population.
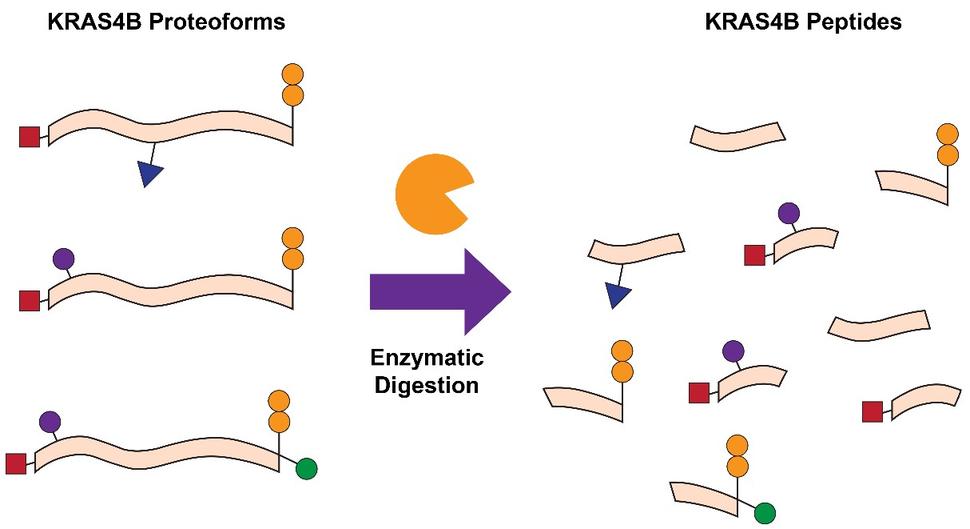
Figure 1. Classical BU proteomic techniques rely on proteolytic digestion of proteins. Resultant peptide masses are then measured in the mass spectrometer.
Another common approach is bottom-up proteomics (BU), which relies on the proteolytic digestion (e.g. trypsin) of proteins, followed by mass determination of the resultant peptides in a mass spectrometer (Fig. 1). BU is frequently applied to determine whether a protein of interest is present in a given sample, quantify the relative or absolute abundance of the protein, and localize key PTMs within the protein sequence. Previous studies have been able to quantify the abundances of RAS isoforms and their mutant variants, examine the impact of KRAS mutations on cellular processes, probe KRAS interactomes, and identify PTMs such as lysine acetylation 2-7. While BU is helpful in these contexts, there is also a significant loss of information that is critical for understanding RAS biology. Due to the high sequence identity (>90%) between the four RAS isoforms, proteolytic digestion leads to the generation of peptides with identical sequences despite originating from distinct RAS isoforms. Furthermore, common RAS mutations occur towards the N-terminal region of the protein, while many of the PTMs which influence function and activity occur towards the C-terminal region. As a result of proteolytic digestion, the connectivity and relationship between mutations and PTMs is lost 8.
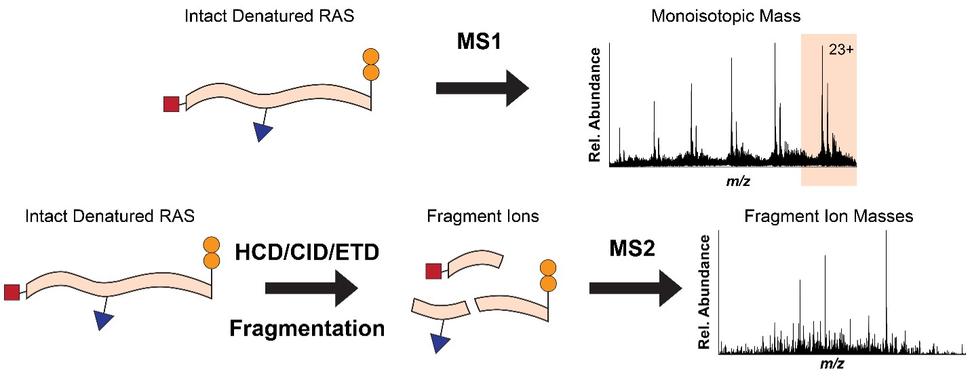
Figure 2. TD proteomics measures the intact monoisotopic proteoform mass before fragmentation within the mass spectrometer.
To overcome the loss of information associated with BU yet maintain the precision of a mass spectrometer, we use top-down proteomics (TD) 8. First, we measure the mass of the intact RAS proteins in the mass spectrometer (MS1). Subsequently, the intact proteins are isolated and subjected to gas bombardment within the mass spectrometer to generate fragment ions (MS2) by a method referred to as higher-energy collisional dissociation (HCD). The masses of these fragment ions are then measured and mapped onto the intact protein sequence, allowing for characterization and localization of PTMs (Fig. 2). By analyzing the intact forms of RAS without any proteolytic digestion, we can understand the combinatorial relationship between different PTMs, mutations, and RAS isoforms. Each of these unique molecular forms of RAS are called proteoforms 9 and can only be elucidated by TD approaches (Fig. 3).
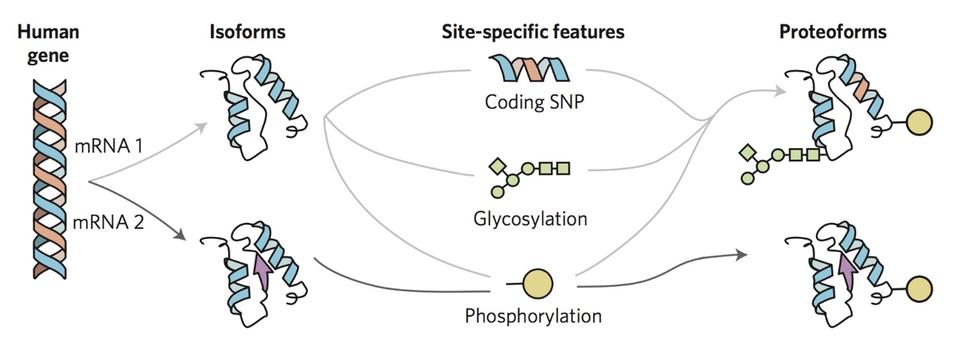
Figure 3. Proteoforms are the different molecular forms in which a protein can be found (i.e. isoforms, SNPs, PTMs, etc.)10
In 2018, the Kelleher laboratory developed an assay combining immunoprecipitation (IP) and TD mass spectrometry (IP-TDMS) to directly isolate and precisely characterize KRAS4B proteoforms in collaboration with the Frederick National Laboratory for Cancer Research (FNLCR) and the Clinical Proteomic Tumor Analysis Consortium (CPTAC). This assay was applied to eight cancer cell lines and six primary colorectal tumors, leading to the identification of eleven KRAS4B proteoforms and two novel PTMs, one of which (Cys118 S-nitrosylation) was revealed to be allele-specific. Additionally, within the six tumor samples analyzed, mutant KRAS4B proteoforms were only detected at low abundance relative to those of wild-type KRAS4B, if detected at all 11. These initial results revealed the potential for proteoform-resolved measurements to illuminate the role of KRAS4B mutations and modifications in human cancer.
Subsequently, we performed substantial method optimization of the IP-TDMS workflow 12, facilitating the detection of additional RAS proteoforms within cancer cell lines. Moreover, we employed the exceptional resolving power and multiple ion fills available on a 21 Tesla FT-ICR mass spectrometer in a collaboration with the National High Magnetic Field Laboratory in Tallahassee, FL, to analyze three isoform-specific MEF cell lines and five cancer cell lines. The ability of this mass spectrometer to increase proteoform signal and therefore dramatically improve detection revealed that the RAS proteoform landscape was far more complicated than we had previously anticipated, indicating that much remains to be understood about the role of these proteoforms in RAS-dependent oncogenic signaling.
Most recently, we applied our optimized RAS IP-TDMS workflow to a panel of twelve cell lines and thirty-four primary colorectal tumors in collaboration with CPTAC and the FNLCR. We first identified and manually validated thirty-nine RAS proteoforms isolated from cell lines, twenty-eight of which had not been previously reported, including two novel classes of RAS proteoforms. In doing so, we observed that KRAS4A proteoforms are present at far lower abundance and exhibit lower PTM diversity than those of KRAS4B, even in cell lines previously determined to express KRAS4A and KRAS4B transcripts at relatively equal abundance 13. Conversely, we observed that KRAS4B wild-type and mutant proteoforms were present at roughly equal abundance, with no significant differences in PTM profile observed between these proteoform populations. These findings underscore the ability of TD to interrogate prominent questions in the RAS field from a new perspective, particularly those covering the importance of KRAS4A in oncogenic signaling 4,5,7,14-16 and the role of KRAS4B PTMs in the previously characterized mutation-specific phenotypes 3,17,18.
We next analyzed a panel of primary colorectal tumors representing seven KRAS mutations (G12C, G12D, G12V, G13D, Q61L, K117N, A145T) from a prior CPTAC study 19 to determine what new information our optimized RAS IP-TDMS assay could provide to complement more conventional methods of tumor sample analysis (e.g. RNA sequencing or BU analysis of phosphorylated peptides). In doing so, we observed that wild-type and mutant KRAS4B proteoforms were present at relatively equal abundance within all respective samples, in contrast to our prior study 11. Even though we did not observe any allele-specific PTMs within the tumor samples, we did identify two novel classes of KRAS4B proteoforms. The first, a KRAS4B proteoform methylated within the C-terminal region, was occasionally observed to be present at higher relative abundance than the canonical KRAS4B proteoform (Cys185 farnesylation and C-terminal carboxymethylation). While the methylation could not be precisely localized to a specific residue due to the high Lys content of the sequence, it may be present on Lys 147, which has been previously reported to be methylated on HRAS 20. The functional relevance of this modification is unknown but may present an interesting avenue for future investigations.
The second novel class of KRAS4B proteoform that we observed within primary tumors comprised of truncated wild-type and mutant KRAS4B proteoforms. These novel proteoforms were detected within over half of the primary tumor samples analyzed and at varying relative abundances with respect to the canonical WT and mutant KRAS4B proteoforms. We performed a control experiment employing a recombinant version of the canonical KRAS4B proteoform developed at the NCI RAS Initiative 21 to demonstrate that the truncation was not due to the IP protocol, sample preparation method, liquid chromatography solvents, or mass spectrometer ionization parameters employed for the IP-TDMS analysis of the primary tumor samples. Given this evidence and the potential significance of the truncated KRAS4B proteoforms, we employed live cell imaging and immunoblotting to further examine its functional relevance. In doing so, we observed that this truncated KRAS4B proteoform exhibited marked differences in membrane localization and MAPK pathway activation compared to the canonical wild-type KRAS4B proteoform. These findings, combined with the wealth of novel RAS proteoforms that we identified within cell lines and tumors, indicate that RAS-dependent signaling pathways may be far more complex and operate by alternative mechanisms than previously thought.
We are currently preparing a manuscript to showcase our recent data, which will soon be submitted for peer review. Our goal for this communication with the RAS community is to bring awareness to the analytical power and novel perspective that top-down proteomics can bring to an established and well-studied field. While there are many benefits of alternative approaches such as immunoblotting and BU proteomics, TD is uniquely positioned to precisely characterize intact proteoforms of RAS. We believe that there are still many open questions regarding RAS biology and RAS-drug interactions that could be addressed in part by TD. Finally, our recent publication in the Ras Activity and Signaling edition of Methods in Molecular Biology offers an in-depth protocol for performing KRAS4B IP-TDMS, so that laboratories outside of the Kelleher Research Group and the NCI RAS Initiative can harness the power of TD to answer their RAS-related questions of interest 12.