Sin1: a novel RAS effector with isoform specificity

Pau Castel, PhD, and Dhirendra Simanshu, PhD
Pau Castel is an Assistant Professor at the Department of Biochemistry and Molecular Biology at New York University School of Medicine. His research is focused on the study of oncoproteins in cancer and genetic disorders. The Castel lab uses biochemical, signal transduction, and mouse genetics to elucidate the function of oncogenic pathways, including RAS and PI3K signaling.
Dhirendra Simanshu leads the structural biology efforts at the NCI RAS Initiative, Frederick National Laboratory for Cancer Research. His research focuses on structural characterization of KRAS in complex with various regulators and targeting druggable pockets using structure-based drug design.
RAS proteins work as molecular binary switches that regulate cellular growth by cycling between inactive GDP- and active GTP-bound states. In humans, three RAS genes encode four different RAS isoforms—HRAS, NRAS, KRAS4A, and KRAS4B—with the last two KRAS isoforms arising from alternative splicing of the fourth KRAS exon. All four RAS isoforms share high sequence and structural homology in the GTPase domain (G-domain; 1–166) and differ significantly in the last 20 amino acids, known as the hypervariable region (HVR), needed for anchoring RAS to the plasma membrane. Upon GTP binding, RAS proteins undergo a conformational change in the two switch regions, termed switch I (residues 30–38) and switch II (amino acids 60–76), which facilitates interaction with various effector proteins resulting in the activation of multiple downstream signaling pathways1. The interaction between RAS-GTP and its effector is mediated by a conserved domain in the effector proteins termed the Ras-binding domain (RBD), or Ras-association domain (RA). More than ten different effector proteins of RAS have been characterized in the last three decades, including well-studied RAF kinases, PI3K, and RALGDS2.
Activating mutations in the RAS genes HRAS, NRAS, and KRAS are frequent in many human tumors, and a group of congenital disorders termed RASopathies3,4. Generally, these mutations promote the GTP-bound state of RAS GTPases and, therefore, associate and activate their downstream effectors. Given the relevance of RAS GTPases in cancer, developmental disorders, and in the fundamental biology of the cell, it is important to identify and characterize all RAS effector proteins. These molecular effectors are responsible for propagating intracellular signaling and contribute to the pathogenesis of RAS mutations, and many pharmacological therapies are being developed to target these critical downstream signaling nodes5.
In this context, we have made some recent efforts in identifying and characterizing proteins that can act as RAS effectors due to the presence of conserved RBD or RA domains. By comprehensively analyzing the domain architecture of the human proteome, we identified a number of proteins that can act as potential RAS effectors. We also focused on those effectors for which the RBD/RA domain was conserved in less complex organisms. One such example is Sin1, which is encoded by the MAPKAP1 gene6. Sin1 was originally identified as a potential RAS effector in a yeast cDNA screening aimed at suppressing the phenotype of the activated RAS ortholog Ras27. Sin1 had also been reported to interact with RAS orthologs in different model organisms8,9.
Sin1 is a protein that contains four recognizable domains, an N-terminal domain (NT), a Conserved Region in the Middle (CRIM) ubiquitin-like fold, a Raf-homology RBD, and a C-terminal pleckstrin homology domain (PH). Sin1 was previously identified as a core component of the mammalian target of rapamycin (mTOR) complex 2 (mTORC2)10–12. This large complex is composed of the kinase mTOR and the regulatory proteins Rictor, mLST8, Protor-1, and Sin1. mTORC2 is a critical cell signaling node because it promotes the phosphorylation and activation of a large number of AGC kinases, which are involved in metabolism, cell growth, and survival13. Sin1 uses its NT to integrate into the Rictor fold and stabilize the mTORC2 complex, as shown by elegant cryoEM studies14. The Sin1 CRIM domain has been shown to participate in the interaction with mTORC2 substrates15, and the PH domain was suggested to interact with phosphoinositides9. Given the important role of Sin1 in the function of mTORC2, we hypothesized that interaction with RAS GTPases through the conserved RBD is important for mTORC2 activity.
First, we confirmed and characterized the interaction between RAS GTPases and Sin1. We found that Sin1 is associated with the GTP-bound G-domain of all canonical RAS GTPases in vitro, with a dissociation constant (KD) ranging from 4 to 5 uM. In contrast, when these experiments were carried out in mammalian cells, Sin1 preferentially binds KRAS4AGTP. Our experiments in cells were undertaken with full-length RAS GTPases as baits, which contained not only the G-domain required for binding, but also the C-terminal HVR which is known to regulate cellular localization and G-domain dynamics16,17. Consistently, we could show that any RAS GTPase containing the KRAS4A HVR had the ability to bind to Sin1 in cells. Interestingly, we found that this HVR does not participate directly in the interaction with Sin1, and it is plausible that the HVR positions the G-domain in an accessible conformation to facilitate Sin1 interaction. Alternatively, the HVR could be responsible for a subcellular localization-specific interaction, although our preliminary data suggests otherwise.
To gain further insights into the structural determinants of Sin1-KRAS interaction, we solved the crystal structure of this complex. In the structure, the KRAS-Sin1 RBD interaction interface resembles the RAS-RAF1 RBD interaction interface18, where the RBD mainly interacts with the KRAS switch-I region. Based on the protein-protein interaction interface observed in the KRAS-Sin1 RBD structure, we generated Sin1 mutants that were likely to disrupt the interaction between Sin1 and KRAS4AGTP, so that we can use them to elucidate the biological function of this interaction in cells. To our surprise, all the mutations in the Sin1 RBD did not affect the interaction in cells. In fact, deletion of the Sin1 RBD yielded a mutant that was still able to partially bind KRAS4AGTP in cells. We hypothesized that Sin1 contains an alternative RAS binding motif that has not been previously reported. To identify such a domain, we decided to undertake an unbiased approach using a technique that we had previously developed termed DoMY-Seq19. DoMY-Seq is a modified yeast two-hybrid technique that uses a saturated library of overlapping cDNA fragments from a protein of interest that are used as prey. Those fragments that interact with the bait can be identified with next-generation sequencing and determine the interacting motif with resolution at the amino acid level. Using this approach, we discovered that the KRAS4A interacting motif of Sin1 did not only include the classical RBD; instead, it also included part of the PH domain that was predicted to fold as an alpha-helix and that we termed the atypical RBD (aRBD). Consistent with these findings, we showed that, in cells, a Sin1 mutant lacking this aRBD was unable to bind KRAS4AGTP. To better understand how this domain mediated interaction with KRAS, we decided to solve the crystal structure of the complex using the Sin1 RBD-PH domain. Interestingly, in the KRAS/Sin1 RBD-PH complex structure, residues located at the N-terminal end of the PH domain interact extensively with the switch II region of KRAS. Overall, our structural work showed that the switch I and II regions of KRAS interact with the RBD and PH domains of Sin1, respectively. The interaction interface formed by the KRAS switch II region in the KRAS/Sin1 RBD-PH complex is much more extensive than any other RAS-effector complexes characterized so far (Figure 1).
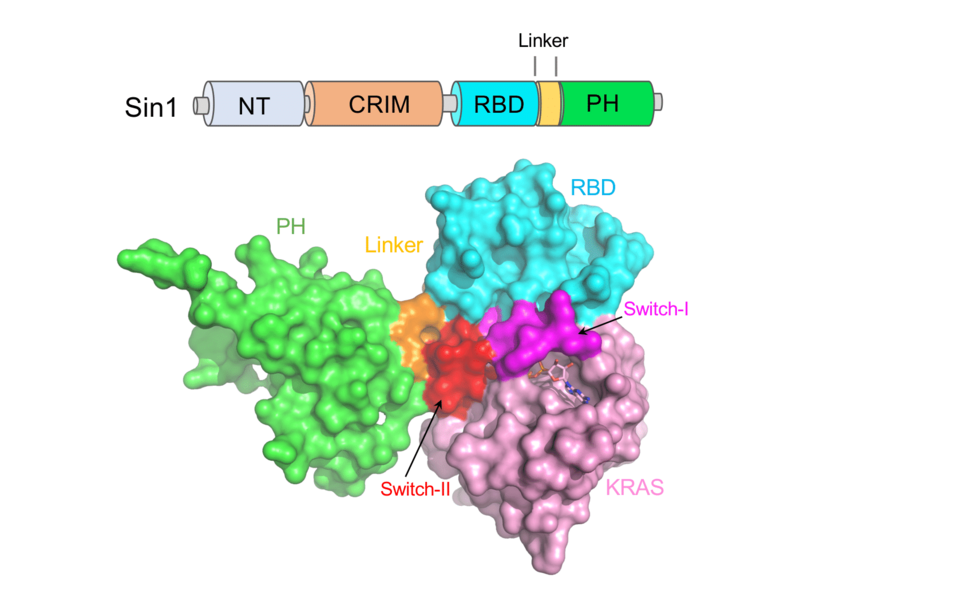
Figure 1: Domain organization of Sin1 and crystal structure of GMPPNP-bound KRAS in complex with Sin1 RBD-PH domains.
With this novel Sin1 mutant (DaRBD) that is unable to bind KRAS, we decided to undertake rescue experiments in cells in which Sin1 had been deleted. Sin1 knockout cells are impaired of mTORC2 activity, as previously shown by others10–12. This can be easily monitored in cells by looking at the phosphorylation of some mTORC2 substrates, such as AKT or PKCa. Sin1 knockout cells can be fully rescued by re-expression of full-length Sin1, but not with a Sin1 mutant that lacks the N-terminal domain required for mTORC2 association. Sin1 DaRBD can completely rescue the mTORC2 phenotype in these cells, indicating that its association with KRAS is dispensable for mTORC2 activity. In addition, we did not detect any changes in mTORC2 assembly and/or composition. We also decided to use this approach at the organismal level. It was previously shown that Sin1 knockout mice are embryonically lethal10; however, the reason for this lethality has not been previously shown. We speculated that this was the result of severe defects in the placenta formation, because Rictor knockout mice display this phenotype20,21. Indeed, we could show that Sin1 knockout mice exhibit fewer maternal spaces and compaction of the trophoblast-lined cords in the placenta. We then leveraged this strong in vivo phenotype to better understand if Sin1 DaRBD would show any defects during embryonic development. We generated mice with a germline in-frame deletion in Sin1 resulting in the DaRBD mutation; these mice were viable and were born at expected Mendelian rates and analysis of their placenta did not show any noticeable defects. Overall, the in vitro and in vivo experiments showed that RAS interaction with Sin1 is not required for modulating mTORC2 activity or assembly.
Although our work provides evidence that RAS interaction is unlikely to control Sin1-mTORC2 function, it also demonstrates that Sin1 is a bona fide KRAS4A effector. There are a few interesting ideas that have emerged from this study:
1. First, if the interaction with KRAS does not affect mTORC2 function, is it possible that Sin1 has a different role in response to KRAS activation? Previous work that was published before the discovery that Sin1 was a key component of mTORC2 proposed that this protein might be a stress-response and described interaction with MEKK2 and JNK22,9.
2. Second, our structural and crosslinking mass spectrometry data suggests that Sin1 is found in an auto-inhibited conformation in which the PH domain occludes the RAS-interacting interface of RBD. If this is the case in cells, what are the molecular triggers that promote the open conformation that allows KRAS binding? A plausible explanation is that lipid binding with the PH domain facilitates the open conformer, although we have been unable to identify such lipid in our preliminary experiments (Figure 2). Other possibilities could include post-translational modifications, i.e., phosphorylation.
3. Lastly, like the RBD and PH domains of Sin1, recent work on tandem domains RBD and CRD of RAF1 have shown that RAS-effector interaction is not limited to RBD/RA domains and switch-I regions of effectors and RAS proteins, respectively. In the last 25 years, most of the biochemical and structural characterizations of the RAS-effector complex have been carried out using the RBD or RA domains of effector proteins. It is possible that domains around the RBD/RA domain in these effector proteins may also play a supporting role in RAS-effector complex formation by interacting with other regions of RAS proteins.
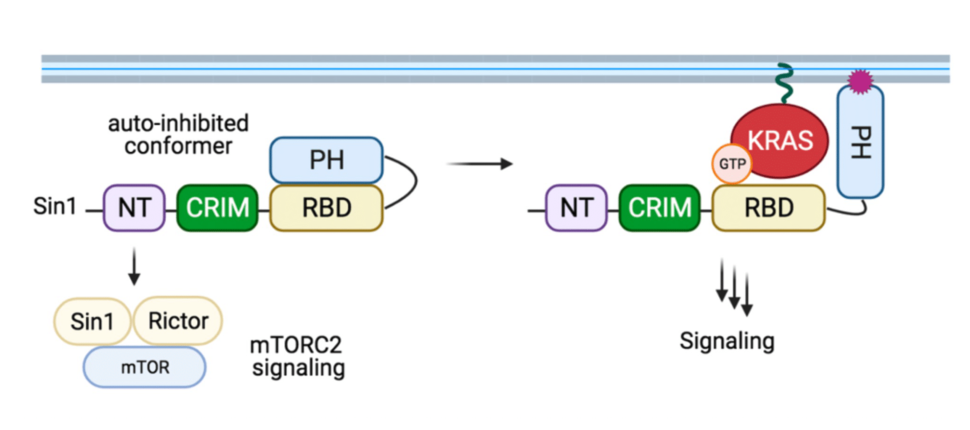
Figure 2: Proposed model of Sin1 function. In the autoinhibited conformation, Sin1 is unable to bind to KRAS but can participate as a component of the mTORC2. We hypothesize that the open conformation results from the interaction with membrane lipids or due to post-translational modification; this conformer is able to bind KRAS-GTP, but the signaling downstream of this GTPase remain unknown.
Overall, we believe that our recent work on Sin1 has revealed novel insights into effector regulation and dynamics, and should inspire similar studies with less-studied RAS effectors. This work will be critical for understanding the signaling specificities of different RAS isoforms and mutants.