Decades of Research Lead to Success
When Brigitte Widemann, M.D., chief of NCI’s Pediatric Oncology Branch (POB), saw selumetinib shrinking the first child’s tumor, she couldn’t believe her eyes.
Then, when she saw the second patient’s tumor shrinking, “I thought, oh my gosh, it’s working!” she recalls. Dr. Widemann has been studying neurofibromatosis type 1 (NF1)—a genetic disease that affects approximately 1 in 3,000 people—since the early 2000s.
Symptoms of NF1 often start in childhood. Up to one-half of kids with NF1 develop slow-growing tumors in nerves, called plexiform neurofibromas (PNs). Although these tumors are not cancerous, they can get very large, compress vital organs, and cause significant pain, disability, and disfigurement. In addition, some PNs turn into a very aggressive cancer called malignant peripheral nerve sheath tumor (MPNST).
Before selumetinib came along, the only treatment for PNs was surgery. But that’s not an option for many patients because the tumors often wrap around nerves, blood vessels, and organs, explained Andrea Gross, M.D., also of POB, part of NCI’s Center for Cancer Research.
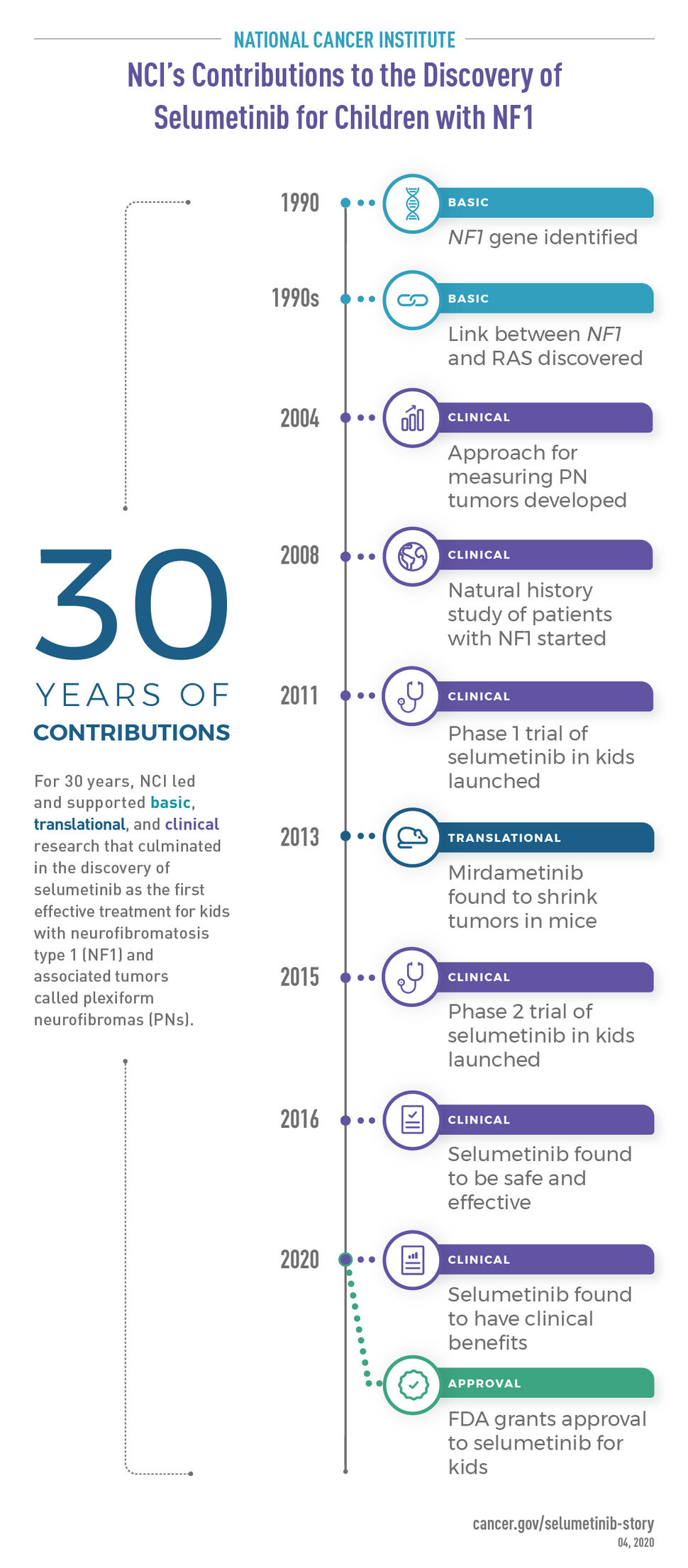
For over 30 years NCI has dedicated substantial time, money, and resources to lead basic, translational, and clinical research of NF1.
“For years POB has served as a hub for clinical trials for the whole NF1 field,” said Jaishri Blakeley, M.D., a clinician scientist with Johns Hopkins University School of Medicine who was involved in studies of selumetinib. “Their expertise, diligence, and commitment have contributed to the vast majority of clinical trials for PN and … [they] have been general leaders in the NF1 field,” she said.
But during those years, “a number of clinical trials did not show the desired results, to the point where I thought we might never find a therapy that works,” Dr. Widemann said.
She and her team persisted, however, and in 2011 launched a small study of selumetinib, sponsored by NCI’s Cancer Therapy Evaluation Program. And this time the therapy worked, shrinking tumors in kids with NF1 and PN.
Later on, a larger study of selumetinib confirmed that the treatment shrinks most PNs and, more importantly, makes children feel better by lessening their pain, physical limitations, and disfigurement. With approval from FDA in April 2020, selumetinib became the first effective treatment for children with NF1 and PNs that can’t be removed by surgery.
"The unique environment of NCI’s intramural program, which enables researchers to dedicate time and resources to developing new treatments for so-called 'rare diseases' such as neurofibromatosis, has contributed greatly to this success, which will have a substantial impact on children with neurofibromatosis," said Douglas Lowy, M.D., NCI’s principal deputy director.
What Causes NF1?
In 1882, a German pathologist named Friedrich von Recklinghausen published a report of an unusual disease and coined its name: neurofibromatosis (“neuro” refers to nerves and “fibroma” means a benign tumor that grows in connective tissues).
Although scientists knew that NF1 was a genetic disease, it wasn’t until 1990 that two independent teams identified the disease-causing gene, aptly called NF1. One team, supported by a grant from NIH, was led by NIH Director Francis Collins, M.D., Ph.D. (then at the University of Michigan). Ray White, Ph.D., then at the University of Utah, led the other team.
Following the discovery of the NF1 gene, scientists set out to understand how mutations in the gene cause the disease. Multiple successive studies established a connection between the protein made by the NF1 gene (called neurofibromin) and RAS, a protein that encourages cell growth and survival. Dr. Lowy made several key contributions, including showing that RAS is overly active in tumor cells from people with NF1.
RAS toggles between “on” and “off” positions, and it turns out that neurofibromin normally helps push RAS into the off position. But when neurofibromin is mutated and not working properly, RAS gets stuck in the on position—causing cells to grow out of control and tumors to form.
With the help of NIH and NCI’s commitment to basic science research, scientists now had a potential target for NF1 treatment: RAS.
Measuring NF1 Tumors
With a target in their sights, Dr. Widemann and her colleagues were eager to start clinical studies of candidate treatments, but first they needed a reliable way to gauge how well the treatments worked.
For most tumors, researchers typically see whether a treatment is working by measuring the diameter of the tumor and seeing if it got smaller. But it is difficult to track changes in the size of PNs using those traditional methods because the tumors form irregular shapes and grow very slowly.
“We realized that we needed better methods to detect changes in tumor size in clinical trials,” Dr. Widemann said.
So in 2004, Jeffrey Solomon, Ph.D., of Sensor Systems, Inc., and NCI scientists developed a reliable and sensitive method of measuring PN tumor volume, called semi-automated volumetric MRI. The technology lets researchers see a change in PN size months or years earlier than is possible with other methods.
NCI researchers, led by Eva Dombi, M.D., of POB, were the first to use semi-automated volumetric MRI to measure treatment response in clinical trials of NF1 treatments. They have since conducted tumor growth evaluations using this method for most NF1 clinical trials in the United States.
A Series of Trials, But No Good Treatments
With a way to measure PNs, the NCI scientists dove headfirst into a series of clinical trials for kids with PNs that can’t be removed by surgery.
Some of the tested treatments targeted RAS or associated “RAS pathways,” while others zeroed in on the cells that surround PN tumors and help them grow. RAS is like a signal controller—it sends out signals that are relayed from protein to protein until the end effect (like cell growth) is accomplished. These signal cascades are called RAS pathways.
The first trial, launched in 2001, compared a placebo to treatment with tipifarnib (Zarnestra), a drug that prevents RAS from being activated. However, tipifarnib did not shrink PNs or delay tumor growth.
Dr. Widemann and her colleagues went on to test a number of other drugs, including pirfenidone (which blocks the growth of cells that make scar tissue and are part of PN tumors), pegylated interferon (which blocks the growth of blood vessels), sorafenib (which targets a protein in a RAS pathway), and sirolimus (which targets a different protein in a RAS pathway).
With the exception of pegylated interferon, none of the tested medicines substantially delayed PN growth or shrank tumors.
“I was at a point where I thought, are we really going to identify something that is going to help our patients?” Dr. Widemann recalled. But “I’ve had patients who participated in several clinical trials that didn’t work, and they didn’t give up, so I didn’t give up…For patients, it’s important to know we are working on it,” she said.
Understanding NF1: The Natural History Study
In the early 2000s, when treatment trials for NF1 had just started, scientists didn’t fully understand how fast NF1 tumors grow and when they grow, and if they could shrink on their own without any treatment.
“Without an understanding of the natural history [of NF1], we wouldn’t be able to design the best trials,” Dr. Widemann said.
So in 2008, NCI scientists launched the Natural History Study of Patients with NF1. Unlike a treatment trial, which looks at the effects of a drug, a natural history study focuses on how a disease progresses on its own. More than 10 years later, the study is still ongoing and is a critical resource for the NF1 research community.
The study looks at growth patterns of PNs and other NF1-related tumors. It also includes assessments of clinical features such as patients’ pain levels, hearing and physical abilities, and cognitive function.
Carrying out such a detailed, long-term study requires years of dedicated funding and extensive collaborations between experts in different specialties that could be found only at NCI, said Dr. Blakeley.
In addition, resources that are available to NCI researchers but would be difficult to access elsewhere, such as whole-body MRI, have been crucial to the study, Dr. Widemann noted.
The natural history study has led to several important advances in NF1 research. For example, the researchers found that PNs grow quickly when patients are young but more slowly during adolescence and young adulthood, and some may actually get smaller when patients are adults. Understanding this pattern has helped researchers choose better measures for clinical trials.
“For trials in adults we can’t use stopping tumor growth as an endpoint, because adult tumors typically do not grow. In contrast, for kids, stopping growth could be a good endpoint,” Dr. Widemann explained.
A Game Changer: Targeting MEK
In 2011, there were still no effective treatments for kids with NF1. The NCI team and their collaborators decided to test selumetinib, a drug that blocks a protein in a RAS pathway called MEK.
Drugs that target MEK are collectively known as MEK inhibitors. Selumetinib was originally developed as a treatment for cancers like melanoma, pancreatic cancer, colorectal cancer, and non-small cell lung cancer.
The phase 1 study, called SPRINT (Selumetinib in Pediatric Neurofibroma Study), enrolled 24 children. To the surprise of the team members, selumetinib shrank the tumors of most patients. It was also relatively safe; the most common side effects were acne-like rash and gastrointestinal problems (such as nausea, vomiting, and diarrhea).
Around the same time, scientists at Cincinnati Children’s Hospital engineered mice that develop tumors similar to PNs. The Cincinnati team, along with NCI researchers and others, worked together to test potential treatments in these mice. In 2013, they found that mirdametinib, another MEK inhibitor, led to “a striking reduction in neurofibroma volumes” in the mice.
This finding was important because there hadn’t been a good way to test drug candidates in mice before, Dr. Widemann explained. This study proved that the mice were a good model of PNs in people and added clout to the idea that a MEK inhibitor might be an effective treatment for PNs.
A Clear Clinical Benefit
Based on the promising results from the phase 1 SPRINT study, in 2015 NCI researchers and their collaborators launched a larger study of selumetinib.
The phase 2 SPRINT study, led by Drs. Gross and Widemann, included patient-reported outcomes pioneered by psychologist Pamela Wolters, Ph.D., of POB. The patient-reported outcomes covered aspects such as patients' pain, physical function, disfigurement, and quality of life.
Selumetinib shrank the tumors of 35 out of 50 participants (70%), and most of the tumors didn’t grow again for over a year. More than half of all participants also reported feeling much better. “They had less pain and felt stronger. Some patients were even able to stop using opioids and other pain medications,” Dr. Gross said.
With all of these data in hand, AstraZeneca—the company that manufactures selumetinib—applied for FDA approval. Data from the NF1 natural history study were a critical component of the FDA approval application because they let the researchers compare the new findings with those from kids who weren’t treated with selumetinib.
“While most patients on the selumetinib trial had tumor shrinkage, most patients on the natural history study had growing tumors. It was clear that selumetinib changed the natural history of NF1 in a good way,” Dr. Widemann explained.
“Had POB not had the vision to carefully and in a longitudinal and systematic fashion collect that natural history data, we would have a much weaker case to present to the FDA and to our patients about the potential value of selumetinib for controlling PN,” Dr. Blakeley said.
“Having that natural history study puts us in a better position to be able to counsel patients about the risks and benefits of being on [selumetinib] versus not,” she added.
More Work to Do
The discovery of selumetinib as an effective treatment for kids with NF1 has opened the door for many other studies of NF1. Even though the approval of selumetinib is a major accomplishment, there is still more work to do to improve outcomes, Dr. Widemann stressed.
“We are very excited and look forward to [NCI’s] continued high-level commitment so that we can make additional progress with our outside collaborators,” she said.
For instance, the NCI team is collaborating with researchers at Indiana University to evaluate different schedules of selumetinib treatment. They hope to reduce the side effects caused by prolonged exposure to the drug.
To better assess the types of pain experienced by people with NF1, Dr. Wolters and Staci Martin, Ph.D., another psychologist in NCI’s POB, are leading a study of updated patient-reported measures of pain intensity and pain interference (how much the pain interferes in the patient’s daily activities). These measures will fill an unmet need for valid and reliable measures of pain in NF1 clinical trials, Dr. Wolters said.
In addition, Dr. Widemann, along with Alice Chen, M.D., and Geraldine O’Sullivan Coyne, M.D., Ph.D., both of NCI’s Developmental Therapeutics Clinic, are testing selumetinib in adults with NF1. Preliminary results from the ongoing trial show that the treatment shrank tumors in the majority of adults and lessened pain.
Findings from the SPRINT studies are also “stimulating new research in multiple directions, not just for PNs,” Dr. Blakeley said. For instance, researchers are looking into using MEK inhibitors to treat people who don't have NF1 but who have cancer with mutations in the NF1 gene, such as some people with melanoma, glioblastoma, or breast cancers.
“As a result of the progress made with selumetinib as the first FDA-approved medicine for children with NF1, there is much optimism that future progress will be achieved more quickly,” Dr. Gross said.
A Major Collaboration
Many different organizations collaborate to ensure that there are no large overlaps or gaps in NF1 research, Dr. Blakeley explained. “The community communicates a lot and supports one another,” she said.
The studies and efforts that led to the approval of selumetinib are the result of collaborations between organizations including:
- NCI’s Cancer Therapy Evaluation Program, which sponsored the SPRINT studies, meaning that they oversaw the trials and provided study monitoring.
- The NIH Clinical Center, Children’s National Hospital in Washington, DC, Children’s Hospital of Philadelphia, and Cincinnati Children’s Hospital, where the SPRINT studies were conducted.
- Neurofibromatosis Therapeutic Acceleration Program (NTAP), a privately funded research program at Johns Hopkins University that is led by Dr. Blakeley and is focused on developing treatments for PNs and NF-related skin tumors. NTAP provided funding to two of the sites involved in the SPRINT trials, enabling the study investigators to enroll 50 patients in less than a year.
- The Children’s Tumor Foundation, a nonprofit organization that funded the mouse studies which identified MEK inhibitors as a potential treatment for PNs and supported the phase I portion of SPRINT.
- REiNS (Response Evaluation in Neurofibromatosis and Schwannomatosis), an international collaboration of scientists that worked together to define useful outcome measures for NF1 clinical studies.
- AstraZeneca, which provided selumetinib and other clinical trial support, and supported studies to understand the drug's distribution in the body (pharmacokinetic studies).
- The Developmental and HyperActive RAS Tumor Specialized Program of Research Excellence (DHART SPORE), an NCI-funded collaborative effort to develop targeted treatments for tumors caused by mutations in the NF1 gene. The SPORE supported studies of selumetinib's chemical interactions in the body (called pharmacodynamics).
- The Neurofibromatosis Clinical Trials Consortium, a consortium sponsored by the Department of Defense that has conducted several clinical trials of children with NF1 and PNs, including trials of other MEK inhibitors.