Targeting Ras Membrane Association: Still an Achilles Heel
, by Mark R. Philips

Dr. Mark Philips.
Oncogenic Ras is a molecular switch stuck in the “on” position. The most direct way of correcting the molecular lesion in mutant Ras would be to devise therapeutics that “unstick” the switch. In an attempt to accomplish this, screens for small molecules that promote GTP hydrolysis were performed at several biopharmaceutical companies in the 1980s but failed to identify any leads1. This outcome was later explained when the co-crystal of Ras and its GTPase activating protein (GAP) revealed that steric hindrance of a critical arginine residue involved in catalysis was the mechanism whereby substitution at glycine 12 activates Ras2. Perhaps the next most direct approach is to find an antagonist of GTP binding. Given the picomolar affinity with which Ras proteins bind guanine nucleotides, a competitive inhibitor seems unlikely. Recently Hunter et al. have breached the affinity barrier with a thiol reactive GDP analog that is able to displace millimolar GDP or GTP, but only in G12C mutants3. A third direct approach to targeting Ras is the search for small molecular weight compounds that bind to the GTPase and thereby block its function, for example by interfering with the ability to interact with effectors. Shima et al. have reported some success along these lines using in silico modeling around the crystal structures of Ras proteins4 and Yan et al. have recently reported success with this approach for Ral GTPases, which are close relatives of Ras5. Steven Fesik and others have taken a fragment-based approach to accomplish this task with some encouraging results6. None of these efforts have led to compounds with KDs below 200 µM. Kevan Shokat recently reported a series of compounds that bind to Ras by virtue of a thiol linkage with cysteine 12 of G12C mutants and thereby perturb the switch regions in a manner that blocks signaling7. Thus, whereas some progress has been made in directly targeting the G domain of the G12C mutant of Ras that is found in ~10% of non-small cell lung cancers, no such success has been reported for the Ras mutants that drive the vast majority of Ras-dependent cancer. The effort to directly target Ras has been the subject of several recent reviews1,8.
In addition to the centrality of the molecular switch mechanism, another tenet of Ras biology is that Ras proteins are peripheral membrane proteins and that association with cellular membranes is absolutely required for their function9,10. If one cannot directly target the guanine nucleotide switch that is Ras, perhaps one can block Ras function by preventing it from getting to the plasma membrane (PM) and other membrane compartments upon which signaling occurs11,12. In other words, tear the broken switch off the wall such that it matters not that it is stuck in the “on” position. This idea gained momentum in 1990 as it became understood how Ras proteins become associated with membranes.
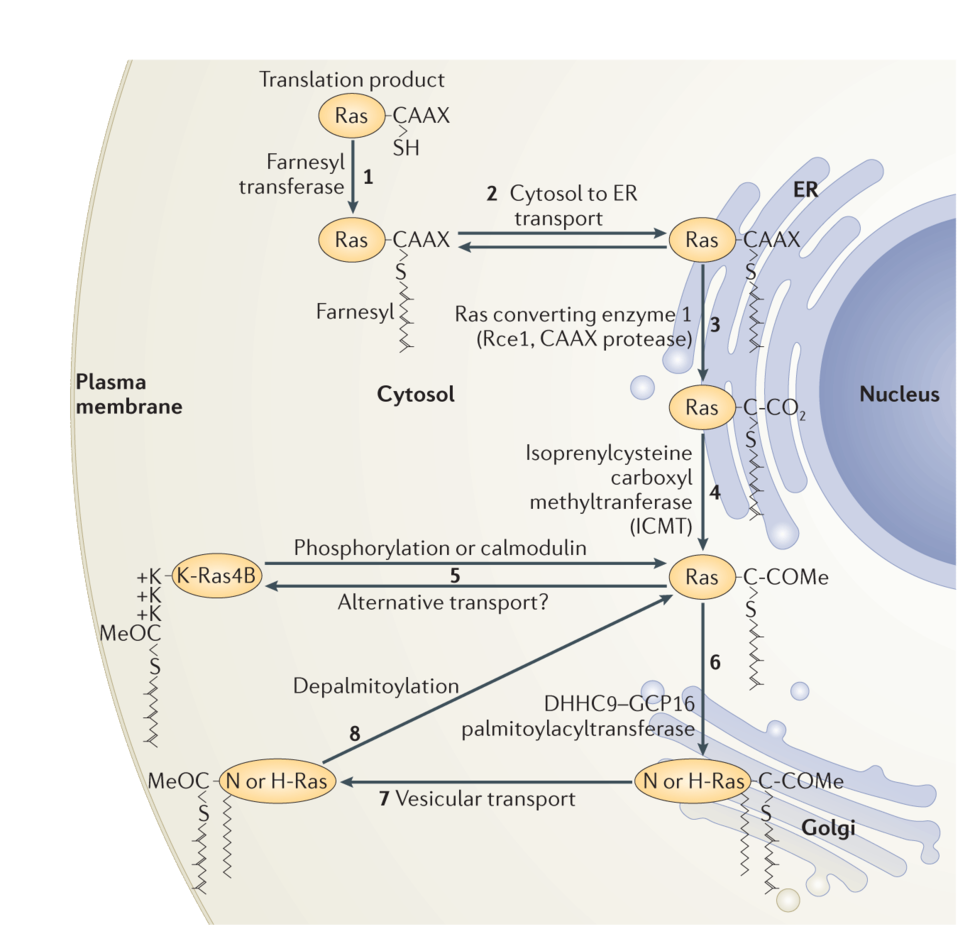
Figure 1. Ras trafficking. Ras is synthesized by cytosolic free polysomes as a globular hydrophilic protein. Nascent Ras encounters farnesyltransferase in the cytosol (1). Once farnesylated, it gains affinity for, and is transported to, membranes of the endoplasmic reticulum (ER) (2) where it encounters the subsequent CAAX processing enzymes Rce1 (3) and Icmt (4). Following CAAX processing, K-Ras4B deviates from the path of the palmitoylated Ras isoforms and proceeds directly to the plasma membrane (5) via a poorly understood pathway that may involve cytosolic chaperones such as PDE6δ. N-Ras and H-Ras proceed to the cytosolic face of the Golgi apparatus where they are palmitoylated by DHHC9–GCP16 and thereby trapped in that membrane compartment (6). From the Golgi they traffic via vesicles to the plasma membrane (7). Upon phosphorylation of serine 181, K-Ras4B can be discharged from the plasma membrane and travel back to the endomembrane system (5). N-Ras and H-Ras are discharged from the membrane by depalmitoylation, and move by retrograde transport back to the Golgi for another round of palmitoylation (8). Like K-Ras4B, N-Ras may be assisted in its transport through the cytosol by PDE6δ. Not shown is K-Ras4A, which has a hybrid targeting motif and is transported independent of PDE6δ.
Reprinted by permission from Macmillan Publishers Ltd: Nat Rev Mol Cell Biol. 2011 Dec 22; 13 (1): 39-51, copyright 2011.
Along with a mating factor of S. cerevisiae, Ras was the founding member of a class of proteins designated CAAX proteins13. These proteins terminate in a CAAX sequence where cysteine is the fourth to last amino acid and is followed by two, usually aliphatic, amino acids and then a widely variable amino acid. Nascent Ras proteins that display a CAAX sequence after release from polysomes become substrates for three enzymes that modify the sequence14. First farnesyl, a 15-carbon polyisoprene lipid, is added to the CAAX cysteine through a stable thioether linkage by farnesyltransferase (FTase). Next the AAX amino acids are removed by a farnesylcysteine-directed endoprotease known as Ras converting enzyme 1 (Rce1). Finally, the carboxyl group of the newly C-terminal farnesylcysteine is methylesterified by isoprenylcysteine carboxylmethyltransferate (Icmt). The end result of this three-step modification of Ras is to convert a globular hydrophilic protein with a hydrophilic C-terminus to one with a hydrophobic C-terminus that has affinity for membranes.
Once the CAAX processing pathway was elucidated and the enzymes that catalyzed the modifications were characterized they became obvious targets for drug discovery. FTase is the first and rate-limiting step of CAAX processing and was therefore the first target in the pathway to be exploited. Aided by CAAX peptide analysis, several pharmaceutical companies developed FTase inhibitors (FTIs) in the early 1990s15. These drugs were orally available, surprisingly non-toxic and hit their target in vivo. Accordingly, one could argue that the development of FTIs was one of the first triumphs of rational drug development. The only problem was that they were ineffective in treating cancer. FTase is one of two CAAX modifying prenyltransferases encoded in eukaryotic genomes. The other, geranylgeranyltransferase I (GGTase I), modifies Ras-related GTPases such as Rho family small GTPases with the C-20 geranylgeranyl polyisoprene lipid. Under normal circumstances, Ras proteins are substrates only for FTase and, in untreated cells, farnesyl is the only polyisoprene that modifies Ras. However, in the presence of FTIs N-Ras and K-Ras, but not H-Ras, become substrates for GGTase I through a process known as alternative prenylation16. Geranylgeranylated Ras proteins associate with membranes and function in a manner identical to their farnesylated versions. Because, when human cancer is driven by mutant Ras, it is almost always driven by mutant K-Ras or N-Ras, alternate prenylation foils FTIs and renders them useless for most cancers. Interestingly, the lack of efficacy of FTIs could not have been predicted from the biology elucidated prior to the development of FTIs because, ironically, the discovery of alternate prenylation required the development of FTIs. Because several pharmaceutical companies invested hundreds of millions of dollars in their FTI programs they viewed the failure of FTIs as a debacle. Consequently, the idea of targeting membrane association of Ras as an approach to anti-cancer therapy fell out of favor for more than a decade. However, if one understands alternative prenylation it is clear that FTIs failed not because blocking Ras association with membranes is a flawed approach but rather because FTIs failed to achieve this goal. Accordingly, we and others have continued to pursue the Ras modification and trafficking pathways as logical targets for potential therapeutics.
Subsequent to the elucidation of CAAX processing it became clear that other factors regulated Ras trafficking and membrane association. In 1990 Hancock17 showed that, in addition to a CAAX sequence, membrane association of Ras proteins requires a “second signal” upstream of the CAAX motif, which comes in two varieties. N-Ras and H-Ras have one or two cysteines in proximity to the CAAX cysteine that are substrates for reversible palmitoylation. Addition of the second class of lipid stabilizes these Ras proteins in membranes. Thus, the first type of second signal is palmitoylation. The best studied of the two splice variants of the KRAS locus, K-Ras4B, has no palmitoylated cysteines but instead possesses a polylysine sequence that participates in an electrostatic interaction with the negatively charged headgroups of the phoshplipids at the inner leaflet of the PM. Thus, the alternate second signal is a polybasic region in proximity to the prenylcysteine.
As originally conceived, Ras proteins were synthesized on free polysomes, modified at their CAAX sequence and, in the case of N-Ras and H-Ras, palmitoylated and then sent directly to the PM as mature, functional signaling molecules. The discovery by Philips that Icmt, one of the CAAX processing enzymes, was restricted to the endoplasmic reticulum (ER)18 forced a reassessment of the Ras trafficking pathway. Rce1 was subsequently also shown to be ER restricted19. Philips20 and Hancock21 showed that, whereas a processed CAAX sequence was capable of delivering proteins to ER membranes, the second signals were required for further trafficking to the PM. Moreover, whereas at steady-state palmitoylated N-Ras and H-Ras localize to both the PM and Golgi apparatus, K-Ras4B was observed only at the PM20. The characterization of Golgi resident DHHC9/GPC16 as the protein acyltransferase (PAT) that modifies N-Ras and H-Ras22 explained the presence of palmitoylated Ras proteins on Golgi where acylation of Ras proteins results in an affinity trap. Recently it has been recognized that delivery of Ras proteins to the PM is not the end of the journey. Rather, they undergo a cycle of delivery to the PM followed by return to endomembrane for recycling. In the case of N-Ras and H-Ras the recycling is initiated by depalmitoylation of Ras at the PM23,24. Interestingly, the prolyl isomerase FKBP12 binds to the membrane proximal region of H-Ras and promotes depalmitoylation25. In the case of K-Ras4B dissociation from the PM is mediated by PKC catalyzed phosphorylation of serine 181 within the polybasic region26, a modification that partially neutralizes the positive charge of the second signal. The current understanding of Ras trafficking is shown in Figure 1.
We have recently determined that K-Ras4A is unique among the four Ras proteins in possessing a dual membrane targeting motif that consists both of a cysteine that becomes palmitoylated and two short polybasic regions flanking the acylated cysteine27. Moreover, unlike K-Ras4B, K-Ras4A does not bind to PDE6δ further differentiating the trafficking of the two splice variants of the KRAS locus27.
Given our dramatically expanded understanding of Ras processing and trafficking what can be done to develop therapeutics that, unlike FTIs, truly interfere with delivery of Ras to cellular membranes? There may still be room to move with regard to CAAX processing. The targets here consist of three enzymes, which lend themselves to inhibitor development. Alternative prenylation explains why FTIs cannot work for K-Ras or N-Ras driven cancers but also suggests that targeting both FTase and GGTase I simultaneously might be successful in blocking oncogenic Ras. Both GGTase I inhibitors (GGTIs) and dual FTase/GGTase I inhibitors have been developed28. Because so many signaling molecules are substrates for one or the other of the prenyltransferases, many cancer biologists and oncologists have considered the potential toxicities of dual inhibition too severe and pharmaceutical companies have been loath to test dual FTase/GGTase inhibitors. But this view does not take into account the real possibility of a therapeutic window for such a drug. Indeed, since the beginning of work on FTIs it has been hoped that prenylation inhibitors will have a greater effect in cancer cells that harbor oncogenic Ras because soluble, GTP-bound Ras can act as a dominant negative by sequestering effectors like Raf-1 (ref. 29) and lead to apoptosis of Ras addicted cells.
Statins that lower plasma cholesterol and reduce the risk of atherosclerosis have been the most prescribed drugs in the US for two decades. This has allowed for robust, retrospective, case-control epidemiologic studies of off target effects, several of which have suggested that these drugs decrease the risk of various cancers including colorectal adenocarcinoma30,31. Statins inhibit 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, which is the first and rate-limiting enzyme in the cholesterol biosynthetic pathway. Because the farnesyl pyrophosphate (FPP) that is an intermediate in cholesterol biosynthetic pathway is also used for the modification of CAAX proteins, it has long been speculated that statins limit cancer by inhibiting prenylation of Ras or related small GTPases. Although in cell culture suprapharmacological levels of statins indeed block protein prenylation and cause mislocalization of Ras32, there is no evidence for this effect in vivo in animals administered pharmacologically relevant doses. FPP is a substrate for both squalene synthase in the cholesterol biosynthetic pathway and FTase in the farnesylation pathway. The KMs of the two enzymes are 2 µM and 5 nM, respectively33, such that FPP levels would have to be lowered 1000 times more to affect protein farnesylation than the decrease required to inhibit cholesterol synthesis, a condition that would require doses of statins unlikely to be tolerated. Thus, there is little rationale for pursuing statins as anti-Ras drugs.
The post-prenylation CAAX processing enzymes are attractive targets for drug development but genetic and pharmacological validation has proven difficult. Knockout of both Rce1 and Icmt are embryonic lethal in mice but Icmt null mice die earlier in gestation than do Rce1 null animals34,35, an observation difficult to explain if these enzymes work sequentially in the CAAX processing pathway. Also perplexing are the observations that, whereas Rce1 deficiency has only modest effects on Ras transformation of rodent fibroblasts36, Icmt deficiency completely blocks contact disinhibited growth of these cells as well as that of cells transformed with BrafV600E (ref. 37). Perhaps most confounding are the observations that, whereas Icmt deficiency ameliorates oncogenic K-Ras driven myeloproliferative disease in a mouse model38, Rce1 deficiency exacerbated the disease39. In an attempt to validate Icmt as a target in a mouse model that better recapitulates human cancer we studied the effect of Icmt deficiency on the Pdx-1-Cre;LSL-K-Ras12D model of pancreatic cancer. Surprisingly, Icmt deficiency exacerbated the disease40. Notch-1 acts as a tumor suppressor in this model41 and further analysis suggested that the reason for the acceleration of the disease was inhibition of Notch signaling as a consequence of Icmt deficiency40. In unpublished work we have found that Icmt deficiency ameliorates K-Ras driven disease in several other mouse models suggesting that the paradoxical findings in the Pdx-1-Cre;LSL-K-Ras12D model is context dependent. These results suggest that we still have much to learn about the biology of CAAX processing.
The complexity of genetic validation of post-prenylation CAAX processing enzymes as targets for anti-cancer drug development not withstanding, effort has gone into producing both Rce1 and Icmt inhibitors. Rce1 inhibitors have been developed and reported33 and in one study such a compound slowed the growth of Ras-transformed cells42. More effort has gone into the development of Icmt inhibitors33,43,44 and the anti-proliferative effects of this class of drug on Ras-transformed cells is more compelling33,44.
Because activating mutations of KRAS drive most human Ras-dependent tumors and because it has been generally accepted that the K-Ras4B splice variant that is not palmitoylated is the major driver, less attention has been paid to pharmacologic inhibition of palmitoylation. However, recent work from the Philips lab implicating the palmitoylated K-Ras4A splice variant in colorectal adenocarcinoma27 may force a re-evaluation of this idea. Moreover, inhibition of palmitoylation may have a place in limiting the activity of oncogenic N-Ras that drives melanoma and hematopoietic malignancies. The idea of non-specific inhibition of protein palmitoylation with compounds like 2-bromopalmitate are problematic since the number of palmitoylated proteins in the human proteome is vast45. Nevertheless, the recent identification of 23 DHHC (Asp-His-His-Cys motif) proteins as the mammalian repertoire of PATs has bolstered the idea of identifying palmitoylation inhibitors that are specific for a subset of substrates including Ras proteins46. However, only limited progress in this area has been reported47.
More progress has been made with inhibitors of depalmitoylation. Although the assignment of acyl protein thioesterase 1 (APT1) as the physiologically relevant enzyme that depalmitoylates Ras remains controversial25,48, the fact that it is a soluble cytosolic enzyme for which a crystal structure is available has facilitated the development of inhibitors46,49,50. Since palmitoylation of Ras targets it to the PM and stabilizes the protein on that compartment where signaling takes place, the idea of inhibiting oncogenic signaling of Ras by blocking depalmitoylation seems somewhat counterintuitive. However, Bastiaens and colleagues have developed an extensive set of observations that suggests that the depalmitoylation on all cellular membranes is required for dynamic cycling of N-Ras and H-Ras on and off membranes and thereby steady-state enrichment of the proteins on the Golgi apparatus and PM51 providing a rationale for inhibiting depalmitoylation as a means of limiting signaling. Indeed, treatment with palmstatin M causes mislocalization of mCitrine-N-Ras to the same endomembrane compartments decorated with mCherry-N-RasC181S that cannot be palmitoylated52.
Aside from CAAX processing and palmitoylation, multiple other post-translational modifications of Ras have been described53 including phosphorylation26, ubiquitination54,55, acetylation56 and nitrosylation57. Whereas ubiquitination, acetylation and nitrosylation have been reported to affect that activation state of the GTPase, phosphorylation26 and ubiquitination54 affect the subcellular localization of the protein. Because each of these modifications is catalyzed by an enzyme, each is, in principle, a target for drug discovery. Perhaps the most intriguing of these post-translational modifications is phosphorylation by PKC of K-Ras4B on serine 181 because it occurs on the isoform most often associated with cancer and because the modification dramatically alters subcellular localization and is associated with decreased survival of transformed cells58. One recent study concluded that phosphorylation of K-Ras4B is required for oncogenesis because rodent fibroblasts transformed with phosphorylation deficient K-Ras4B12D,181A failed to produce tumors in nude mice59, a result that contradicted previous reports of the same experiment26. We have found that mice with an LSL-K-Ras4B,12D,181A double knock-in allele produced pancreatic tumors when crossed with P48-Cre transgenic animals with equal frequency as mice with an LSL-K-Ras4B,12D,181S allele (unpublished results), arguing strongly against a requirement for K-Ras4B phosphorylation. Perhaps most compelling with regard to potential therapeutics is the observation that bryostatin, a PKC agonist, slowed the growth of tumors driven by oncogenic K-Ras4B but not those driven by a phosphorylation-deficient mutant of K-Ras4B26. However, enthusiasm for this approach to cancer therapy is somewhat diminished by the requirement to find a drug that stimulates phosphorylation of K-Ras4B without affecting PKC mediated activation of other signaling molecules that promote tumor growth and/or lead to toxicities.
Whereas some aspects of Ras trafficking involve vesicular transport, photobleaching and photoactivation studies of Ras tagged with fluorescent proteins have established that Ras proteins can be delivered from one membrane compartment to another rapidly by diffusion through the cytosol23,24. This raises the question of how the farnesyl lipid is shielded from the aqueous environment of the cytosol. RhoGDI, which forms complexes with Rho, Rac and Cdc42, is the best characterized example of a prenylprotein binding protein that can carry small GTPases through the cytosol60,61. Although several proteins that bind to Ras in a farnesyl-dependent fashion have been described62, including PRA1 (ref. 63), galectin 1 (ref. 64) and smgGDS65, until recently it was not clear that a RhoGDI-like chaperone is required by Ras. Not only is it now well established that the δ subunit of cGMP phosphodiesterase type 6 (PDE6δ) binds to farnesylated Ras in the cytosol66-68 but Wittinghoffer and colleagues have provided insight into how release of bound Ras is regulated by GTP-bound Arl2/3 (ref. 69). Taking advantage of Wittinghoffer’s co-crystal of PDE6δ and the farnesylated Ras-related GTPase Rheb69, Waldmann and colleagues have produced inhibitors of the PDE6δ-Ras interaction that alter the localization of K-Ras4B and slow the growth of tumor cells70,71. Because PDE6δ binds K-Ras4B but not K-Ras4A27, it is not clear what will be the efficacy of anti-PDE6δ drugs since tumors that harbor a mutant KRAS gene express oncogenic forms of both splice variants27. Thus, although more studies are needed with regard to PDE6δ inhibitors, interfering with cytosolic chaperones may represent yet another approach to inhibiting oncogenic Ras by blocking subcellular trafficking.
The enormous explosion in the understanding of how nascent Ras proteins are delivered to the PM that has occurred over the past 25 years has revealed many targets for rational drug design. However, it is likely that the Ras trafficking pathway has not been fully revealed and that other proteins play a role in membrane targeting and/or stabilization. Several groups have taken an unbiased approach to identify inhibitors of Ras trafficking and new genes involved in the pathway that might prove good targets for drug discovery. Hancock and colleagues devised a high-content screening assay that measures mislocalization of K-Ras4B from PM to endomembrane and identified fendiline, an L type calcium channel blocker, as a compound that induces K-Ras4B mislocalization72. Visual screens like the one employed by Hancock are difficult to standardize and quantify. In the Philips lab Nicole Fehrenbacher has devised a dual luciferase assay that reports displacement of K-Ras4B from cellular membranes and the assay is currently being employed in a genome-wide siRNA screen designed to identify new regulators of K-Ras4B membrane association.
In conclusion, although the failure of FTIs two decades ago was a real setback in the field, the Ras trafficking pathway remains a logical, validated and viable approach for the development of anti-Ras drugs. Indeed the greatly expanded understanding of Ras trafficking has provided a target-rich environment for drug discovery.
Selected References
- Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging Ras back in the ring. Cancer Cell. 2014; 25: 272-81.
- Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, Wittinghofer A. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic mutants. Science. 1997; 277: 5324.
- Hunter JC, Gurbani D, Ficarro SB, Carrasco MA, Lim SM, Choi HG, Xie T, Marto JA, Chen Z, Gray NS, Westover KD. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc Natl Acad Sci U S A. 2014; 111: 8895-900.
- Shima F, Yoshikawa Y, Ye M, Araki M, Matsumoto S, Liao J, Hu L, Sugimoto T, Ijiri Y, Takeda A, Nishiyama Y, Sato C, Muraoka S, Tamura A, Osoda T, Tsuda K, Miyakawa T, Fukunishi H, Shimada J, Kumasaka T, Yamamoto M, Kataoka T. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc Natl Acad Sci U S A. 2013; 110: 8182-7.
- Yan C, Liu D, Li L, Wempe MF, Guin S, Khanna M, Meier J, Hoffman B, Owens C, Wysoczynski CL, Nitz MD, Knabe WE, Ahmed M, Brautigan DL, Paschal BM, Schwartz MA, Jones DN, Ross D, Meroueh SO, Theodorescu D. Discovery and characterization of small molecules that target the GTPase Ral. Nature. 2014; 515: 443-7.
- Sun Q, Burke JP, Phan J, Burns MC, Olejniczak ET, Waterson AG, Lee T, Rossanese OW, Fesik SW. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew Chem Int Ed Engl. 2012; 51: 6140-3.
- Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013; 503: 548-51.
- Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission Possible? Nature Reviews Drug Discovery. 2014; 13: 828-51.
- Willumsen BM, Christensen A, Hubbert NL, Papageorge AG, Lowy DR. The p21 Ras C-terminus is required for transformation and membrane association. Nature. 1984; 310: 583-6.
- Jackson JH, Cochrane CG, Bourne JR, Solski PA, Buss JE, Der CJ. Farnesol modification of Kirsten-ras exon 4B protein is essential for transformation. Proc Natl Acad Sci U S A. 1990; 87: 3042-6.
- Chiu VK, Bivona T, Hach A, Sajous JB, Silletti J, Wiener H, Johnson RL, Cox AD, Philips MR. Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol. 2002; 4: 343-50.
- Bivona TG, Perez De Castro I, Ahearn IM, Grana TM, Chiu VK, Lockyer PJ, Cullen PJ, Pellicer A, Cox AD, Philips MR. Phospholipase Cgamma activates Ras on the Golgi apparatus by means of RasGRP1. Nature. 2003; 424: 694-8.
- Lowy DR, Willumsen BM. Protein modification: new clue to Ras lipid glue. Nature. 1989; 341: 384-5.
- Wright LP, Philips MR. Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J Lipid Res. 2006; 47: 883-91.
- Cox AD, Der CJ. Farnesyltransferase inhibitors: promises and realities. Curr Opin Pharmacol. 2002; 2: 388-93.
- Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, Bishop WR, Pai JK. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997; 272: 14459-64.
- Hancock JF, Paterson H, Marshall CJ. A polybasic domain or palmitoylation is required in addition to the CAAX motif to loacalize p21ras to the plasma membrane. Cell. 1990; 63: 133-9.
- Dai Q, Choy E, Chiu V, Romano J, Slivka S, Steitz S, Michaelis S, Philips MR. Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. Journal of Biological Chemistry. 1998; 273: 15030-4.
- Schmidt WK, Tam A, Fujimura-Kamada K, Michaelis S. Endoplasmic reticulum membrane localization of Rce1p and Ste24p, yeast proteases involved in carboxyl-terminal CAAX protein processing and amino-terminal a-factor cleavage. Proceedings of the National Academy of Sciences (USA). 1998; 95: 11175-80.
- Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, Ivanov IE, Philips MR. Endomembrane trafficking of Ras: the CAAX motif targets proteins to the ER and Golgi. Cell. 1999; 98: 69-80.
- Apolloni A, Prior IA, Lindsay M, Parton RG, Hancock JF. H-Ras but not K-Ras traffics to the plasma membrane through the exocytic pathway. Mol Cell Biol. 2000; 20: 2475-87.
- Swarthout JT, Lobo S, Farh L, Croke MR, Greentree WK, Deschenes RJ, Linder ME. DHHC9 and GCP16 constitute a human protein fatty acyltransferase with specificity for H- and N-Ras. J Biol Chem. 2005; 280: 31141-8.
- Rocks O, Peyker A, Kahms M, Verveer PJ, Koerner C, Lumbierres M, Kuhlmann J, Waldmann H, Wittinghofer A, Bastiaens PI. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005; 307: 1746-52.
- Goodwin JS, Drake KR, Rogers C, Wright L, Lippincott-Schwartz J, Philips MR, Kenworthy AK. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J Cell Biol. 2005; 170: 261-72.
- Ahearn IM, Tsai FD, Court H, Zhou M, Jennings BC, Ahmed M, Fehrenbacher N, Linder ME, Philips MR. FKBP12 binds to acylated H-Ras and promotes depalmitoylation. Mol Cell. 2011; 41: 173-85.
- Bivona TG, Quatela SE, Bodemann BO, Ahearn IO, Soskis MJ, Mor A, Miura J, Wiener HH, Wright L, Saba SG, Yim D, Fein A, Perez de Castro I, Li C, Thompson CB, Cox AD, Philips MR. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006; 21: 481-93.
- Tsai FD, Lopes MS, Zhou M, Court H, Ponce O, Fiordalisi JJ, Gierut JJ, Cox AD, Haigis KM, Philips MR. K-Ras4A splice variant is widely expressed in cancer and
uses a hybrid membrane-targeting motif. Proc Natl Acad Sci U S A. 2015 Jan 5. pii: 201412811. [Epub ahead of print] PubMed PMID: 25561545. - Sebti SM, Hamilton AD. Farnesyltransferase and geranylgeranyltransferase I inhibitors and cancer therapy: lessons from mechanism and bench-to-bedside translational studies. Oncogene. 2000; 19: 6584-93.
- Fiordalisi JJ, Holly SP, Johnson RL, 2nd, Parise LV, Cox AD. A distinct class of dominant negative Ras mutants: cytosolic GTP-bound Ras effector domain mutants that inhibit Ras signaling and transformation and enhance cell adhesion. J Biol Chem. 2002; 277: 10813-23.
- Poynter JN, Gruber SB, Higgins PD, Almog R, Bonner JD, Rennert HS, Low M, Greenson JK, Rennert G. Statins and the risk of colorectal cancer. N Engl J Med. 2005; 352: 2184-92.
- Graaf MR, Richel DJ, van Noorden CJ, Guchelaar HJ. Effects of statins and farnesyltransferase inhibitors on the development and progression of cancer. Cancer Treatment Reviews. 2004; 30: 609-41.
- Cho KJ, Hill MM, Chigurupati S, Du G, Parton RG, Hancock JF. Therapeutic levels of the hydroxmethylglutaryl-coenzyme A reductase inhibitor lovastatin activate Ras signaling via phospholipase D2. Mol Cell Biol. 2011; 31: 1110-20.
- Winter-Vann AM, Casey PJ. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat Rev Cancer. 2005; 5: 405-12.
- Bergo MO, Leung GK, Ambroziak P, Otto JC, Casey PJ, Gomes AQ, Seabra MC, Young SG. Isoprenylcysteine carboxyl methyltransferase deficiency in mice. J Biol Chem. 2001; 276: 5841-5.
- Lin X, Jung J, Kang D, Xu B, Zaret KS, Zoghbi H. Prenylcysteine carboxylmethyltransferase is essential for the earliest stages of liver development in mice. Gastroenterology. 2002; 123: 345-51.
- Bergo MO, Ambroziak P, Gregory C, George A, Otto JC, Kim E, Nagase H, Casey PJ, Balmain A, Young SG. Absence of the CAAX endoprotease Rce1: effects on cell growth and transformation. Mol Cell Biol. 2002; 22: 171-81.
- Bergo MO, Gavino BJ, Hong C, Beigneux AP, McMahon M, Casey PJ, Young SG. Inactivation of Icmt inhibits transformation by oncogenic K-Ras and B-Raf. Journal of Clinical Investigation. 2004; 113: 539-50.
- Wahlstrom AM, Cutts BA, Liu M, Lindskog A, Karlsson C, Sjogren AK, Andersson KM, Young SG, Bergo MO. Inactivating Icmt ameliorates K-RAS-induced myeloproliferative disease. Blood. 2008; 112: 1357-65.
- Wahlstrom AM, Cutts BA, Karlsson C, Andersson KM, Liu M, Sjogren AK, Swolin B, Young SG, Bergo MO. Rce1 deficiency accelerates the development of K-RAS-induced myeloproliferative disease. Blood. 2007; 109: 763-8.
- Court H, Amoyel M, Hackman M, Lee KE, Xu R, Miller G, Bar-Sagi D, Bach EA, Bergo MO, Philips MR. Isoprenylcysteine carboxylmethyltransferase deficiency exacerbates KRAS-driven pancreatic neoplasia via Notch suppression. J Clin Invest. 2013; 123: 4681-94.
- Hanlon L, Avila JL, Demarest RM, Troutman S, Allen M, Ratti F, Rustgi AK, Stanger BZ, Radtke F, Adsay V, Long F, Capobianco AJ, Kissil JL. Notch1 functions as a tumor suppressor in a model of K-Ras-induced pancreatic ductal adenocarcinoma. Cancer Res. 2010; 70: 4280-6.
- Chen Y. Selective inhibition of Ras-transformed cell growth by a novel fatty acid-based chloromethyl ketone designed to target Ras endoprotease. Annals of the New York Academy of Sciences. 1999; 886: 103-8.
- Judd WR, Slattum PM, Hoang KC, Bhoite L, Valppu L, Alberts G, Brown B, Roth B, Ostanin K, Huang L, Wettstein D, Richards B, Willardsen JA. Discovery and SAR of methylated tetrahydropyranyl derivatives as inhibitors of isoprenylcysteine carboxyl methyltransferase (ICMT). J Med Chem. 2011; 54: 5031-47.
- Lau HY, Ramanujulu PM, Guo D, Yang T, Wirawan M, Casey PJ, Go ML, Wang M. An improved isoprenylcysteine carboxylmethyltransferase inhibitor induces cancer cell death and attenuates tumor growth in vivo. Cancer Biol Ther. 2014; 15: 1280-91.
- Yang W, Di Vizio D, Kirchner M, Steen H, Freeman MR. Proteome scale characterization of human S-acylated proteins in lipid raft-enriched and non-raft membranes. Mol Cell Proteomics. 2010; 9: 54-70.
- Chavda B, Arnott JA, Planey SL. Targeting protein palmitoylation: selective inhibitors and implications in disease. Expert Opinion on Drug Discovery. 2014; 9: 1005-19.
- Ducker CE, Griffel LK, Smith RA, Keller SN, Zhuang Y, Xia Z, Diller JD, Smith CD. Discovery and characterization of inhibitors of human palmitoyl acyltransferases. Mol Cancer Ther. 2006; 5: 1647-59.
- Duncan JA, Gilman AG. Characterization of Saccharomyces cerevisiae acyl-protein thioesterase 1, the enzyme responsible for G protein alpha subunit deacylation in vivo. J Biol Chem. 2002; 277: 31740-52.
- Dekker FJ, Rocks O, Vartak N, Menninger S, Hedberg C, Balamurugan R, Wetzel S, Renner S, Gerauer M, Scholermann B, Rusch M, Kramer JW, Rauh D, Coates GW, Brunsveld L, Bastiaens PI, Waldmann H. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat Chem Biol. 2010; 6: 449-56.
- Zimmermann TJ, Burger M, Tashiro E, Kondoh Y, Martinez NE, Gormer K, Rosin-Steiner S, Shimizu T, Ozaki S, Mikoshiba K, Watanabe N, Hall D, Vetter IR, Osada H, Hedberg C, Waldmann H. Boron-based inhibitors of acyl protein thioesterases 1 and 2. Chembiochem. 2013; 14: 115-22.
- Rocks O, Gerauer M, Vartak N, Koch S, Huang ZP, Pechlivanis M, Kuhlmann J, Brunsveld L, Chandra A, Ellinger B, Waldmann H, Bastiaens PI. The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell. 2010; 141: 458-71.
- Hedberg C, Dekker FJ, Rusch M, Renner S, Wetzel S, Vartak N, Gerding-Reimers C, Bon RS, Bastiaens PIH, Waldmann H. Development of highly potent inhibitors of the Ras-targeting human acyl protein thioesterases based on substrate similarity design. Angewandte Chemie International Ed In English. 2011; 50: 9832-7.
- Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol. 2012; 13: 39-51.
- Jura N, Scotto-Lavino E, Sobczyk A, Bar-Sagi D. Differential modification of Ras proteins by ubiquitination. Mol Cell. 2006; 21: 679-87.
- Sasaki AT, Carracedo A, Locasale JW, Anastasiou D, Takeuchi K, Kahoud ER, Haviv S, Asara JM, Pandolfi PP, Cantley LC. Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci Signal. 2010; 4: ra13.
- Yang MH, Nickerson S, Kim ET, Liot C, Laurent G, Spang R, Philips MR, Shan Y, Shaw DE, Bar-Sagi D, Haigis MC, Haigis KM. Regulation of RAS oncogenicity by acetylation. Proc Natl Acad Sci U S A. 2012; 109: 10843-8.
- Williams JG, Pappu K, Campbell SL. Structural and biochemical studies of p21Ras S-nitrosylation and nitric oxide-mediated guanine nucleotide exchange. Proc Natl Acad Sci U S A. 2003; 100: 6376-81.
- Sung PJ, Tsai FD, Vais H, Court H, Yang J, Fehrenbacher N, Foskett JK, Philips MR. Phosphorylated K-Ras limits cell survival by blocking Bcl-xL sensitization of inositol trisphosphate receptors. Proc Natl Acad Sci U S A. 2013; 110: 20593-8.
- Barcelo C, Paco N, Morell M, Alvarez-Moya B, Bota-Rabassedas N, Jaumot M, Vilardell F, Capella G, Agell N. Phosphorylation at Ser-181 of oncogenic KRAS is required for tumor growth. Cancer Res. 2014; 74: 1190-9.
- Hoffman GR, Nassar N, Cerione RA. Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell. 2000; 100: 345-56.
- Dovas A, Couchman JR. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem J. 2005; 390: 1-9.
- Kloog Y, Cox AD. Prenyl-binding domains: potential targets for Ras inhibitors and anti-cancer drugs. Seminars in cancer biology. 2004; 14: 253-61.
- Figueroa C, Taylor J, Vojtek AB. Prenylated Rab acceptor protein is a receptor for prenylated small GTPases. J Biol Chem. 2001; 276: 28219-25.
- Rotblat B, Niv H, Andre S, Kaltner H, Gabius HJ, Kloog Y. Galectin-1(L11A) predicted from a computed galectin-1 farnesyl-binding pocket selectively inhibits Ras-GTP. Cancer Res. 2004; 64: 3112-8.
- Berg TJ, Gastonguay AJ, Lorimer EL, Kuhnmuench JR, Li R, Fields AP, Williams CL. Splice variants of SmgGDS control small GTPase prenylation and membrane localization. J Biol Chem. 2010; 285: 35255-66.
- Nancy V, Callebaut I, El Marjou A, de Gunzburg J. The delta subunit of retinal rod cGMP phosphodiesterase regulates the membrane association of Ras and Rap GTPases. J Biol Chem. 2002; 277: 15076-84.
- Chandra A, Grecco HE, Pisupati V, Perera D, Cassidy L, Skoulidis F, Ismail SA, Hedberg C, Hanzal-Bayer M, Venkitaraman AR, Wittinghofer A, Bastiaens PI. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat Cell Biol. 2012; 14: 148-58.
- Philips MR. Ras hitchhikes on PDE6delta. Nat Cell Biol. 2012; 14: 128-9.
- Ismail SA, Chen Y-X, Rusinova A, Chandra A, Bierbaum M, Gremer L, Triola G, Waldmann H, Bastiaens PIH, Wittinghofer A. Arl2-GTP and Arl3-GTP regulate a GDI-like transport system for farnesylated cargo. Nature Chemical Biology. 2011; 7: 942-9.
- Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens PIH, Waldmann H. Small molecule inhibition of the KRAS-PDEd interaction impairs oncogenic KRAS signalling. Nature. 2013; 497: 638-42.
- Zimmermann G, Schultz-Fademrecht C, Kuchler P, Murarka S, Ismail S, Triola G, Nussbaumer P, Wittinghofer A, Waldmann H. Structure guided design and kinetic analysis of highly potent benzimidazole inhibitors targeting the PDEdelta prenyl binding site. J Med Chem. 2014; 57: 5435-48.
- van der Hoeven D, Cho KJ, Ma X, Chigurupati S, Parton RG, Hancock JF. Fendiline inhibits K-Ras plasma membrane localization and blocks K-Ras signal transmission. Mol Cell Biol. 2013; 33: 237-51.