Ras Mitogenic Signaling: p53 Enters the Stage
, by Matthias Drosten and Mariano Barbacid

Dr. Mathias Drosten and Prof. Mariano Barbacid
Ras proteins and their downstream signaling pathways have been studied for more than 40 years1. Soon after their discovery and molecular characterization, it became evident that Ras proteins are directly implicated in the control of cell proliferation. Nevertheless, our understanding of the molecular mechanisms by which Ras proteins convey mitogenic signals to the cell cycle machinery remains incomplete.
The immediate events that regulate Ras activation are reasonably well known. Likewise, there is ample information regarding their immediate effectors, in particular the members of the MAPK signaling cascade, the Raf, Mek and Erk kinases1,2. Indeed, genetic interrogation of these effectors has indicated that these kinases are solely responsible for mediating mitogenic signaling at least in mouse embryonic fibroblasts (MEFs) or cultured keratinocytes3,4. However, the precise mechanism of how this information is transmitted to the nuclear machinery to control cell proliferation remains mostly unknown. Here, we recapitulate our recent studies aimed at identifying some of these missing links using an unbiased genetic approach. We also intend to discuss potential implications for cancer therapy.
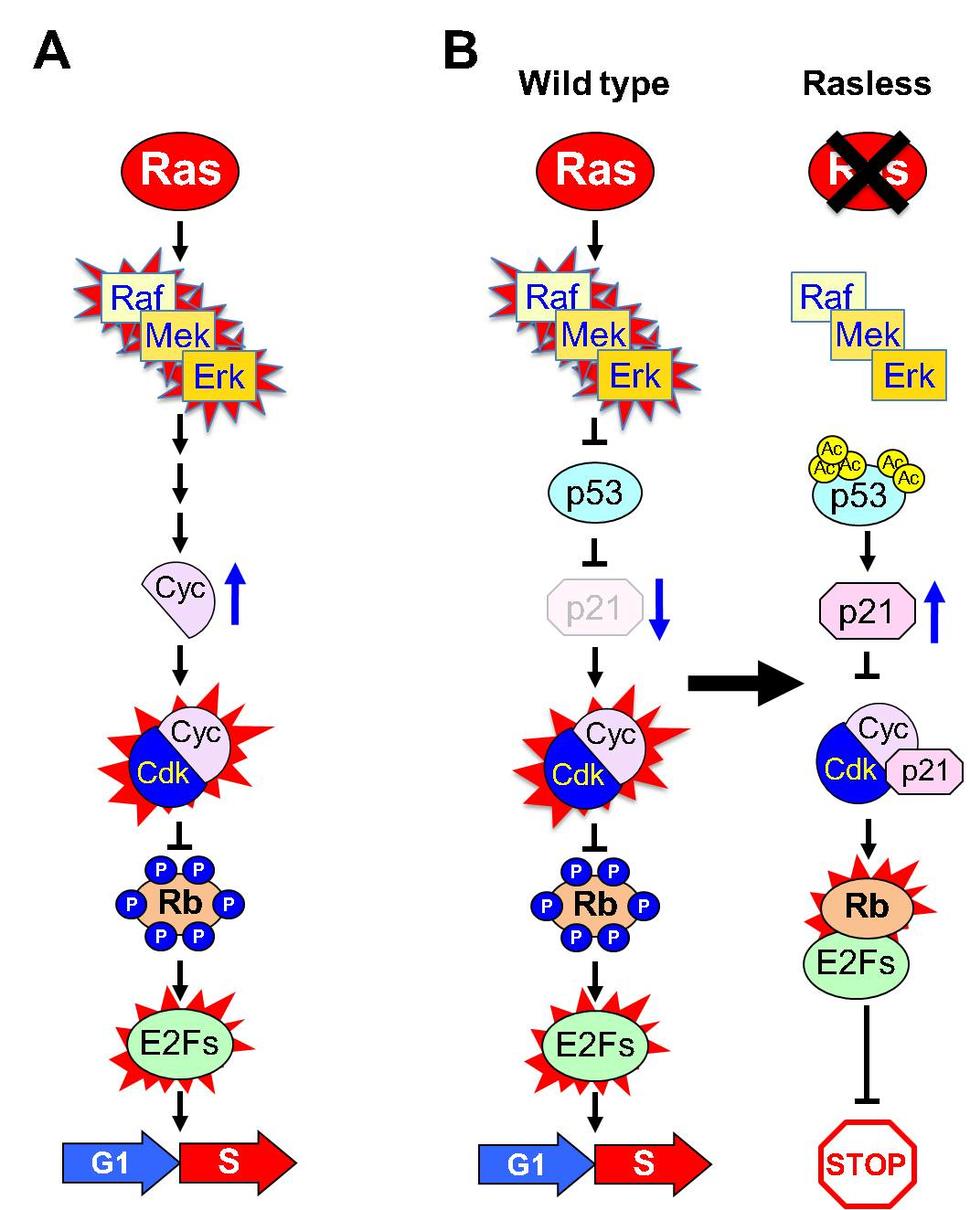
FIGURE 1: (A) Schematic representation of current model of the mitogenic Ras signaling pathway. Black arrows indicate increased activity, T lines represent decreased activity. Upward and downward blue arrows represent increased or decreased amounts of protein. Red spikes around protein symbols represent the active states of those proteins. Ras activates Raf, Mek, and Erk which through transcriptional mechanisms increase the amount of cyclins (Cyc). Increased cyclin levels enhance the activity of cyclin-dependent kinases (Cdk), resulting in increased phosphorylation of Rb, thereby releasing E2F transcription factors to transcribe genes necessary for progression of the cell cycle from G1 to S. (B) Modified representation of this pathway in wild type (left) and Rasless (right) MEFs. Importantly, we have shown previously (reference 3) that levels of cyclins bound to Cdks remain normal in the absence of Ras. Our results instead indicate that levels of the cyclin/Cdk inhibitor p21 protein are critical. The left panel shows how activated Raf, Mek, and Erk stimulate the activity of E2Fs by decreasing the activity of p53, thus decreasing expression of p21. In cells lacking all Ras genes (right panel) absence of activated Raf, Mek, and Erk causes acetylation of p53, thereby increasing expression of p21, inhibition of the cyclin/Cdk kinase activity, increased hypophosphorylated Rb, and increased sequestration of E2F activity into inactive complexes with Rb. Lower levels of active E2Fs stall cell cycling.
As expected, genetic ablation of the three Ras proteins (H-, N- and K-Ras) in MEFs and keratinocytes completely prevents cell proliferation3,4. Yet, ectopic expression of activated forms of the main components of the MAPK pathway efficiently restored their proliferative capacity, thus demonstrating the reversible nature of the quiescent Rasless state. Earlier genetic studies demonstrated that the retinoblastoma protein (Rb) was the ultimate target of active Ras signaling in cell cycle control5. Indeed, knock down as well as inactivation of Rb licensed quiescent Rasless cells to enter the cell cycle3. In this regard, it was proposed that Ras proteins controlled Rb activity by inducing expression of D- and E-type cyclins, which after binding to their catalytic partners, Cdk4/6 and Cdk2, led to Rb phosphorylation and resultant inactivation of E2F binding6 (Figure 1A). This hypothesis, however, needs to be revised since as we have recently shown, Rasless cells retain normal levels of D- and E-type cyclins bound to their cognate Cdks3. Yet, these cyclin/Cdk complexes lack substantial kinase activity suggesting that Ras signaling is essential to control their enzymatic activity, presumably via their interaction with Cdk inhibitors3 (Figure 1B)
To get insights into how Ras proteins regulate cell proliferation, we performed an unbiased small hairpin RNA (shRNA) library screen in Rasless cells. This screen unambiguously identified the p53/p21Cip1 axis as an essential mediator of Ras mitogenic signaling7. That is, efficient knock down of either p53 or p21Cip1 expression fully restored the proliferative properties of Rasless cells (Figure 2A). Subsequent studies revealed that Ras signaling had no effect on p53 expression levels but completely inhibited p21Cip1 expression7. Interestingly, knock down of any of the members of the INK4 family of Cdk inhibitors, including p16INK4a or p15INK4b, could not rescue Rasless cells from their quiescent state, indicating that these cell cycle regulators are not directly involved in Ras mitogenic signaling as previously proposed.
How does Ras signaling control p21Cip1 expression levels? Further studies revealed that loss of Ras proteins caused widespread transcriptional activation of p53 through a mechanism involving acetylation of specific residues in its DNA binding domain7. Surprisingly, phosphorylation and/or protein stabilization of p53 was not required. Knock down of other well-known p53 regulated genes, including Gadd45a, Pai-1 or Puma had no effect on Ras-mediated mitogenic signaling. Thus, these genetic studies place the p53/p21 axis at the center of the mitogenic pathway that connects Ras signaling with the cell cycle (Figure 1B). However, the precise mechanism linking active Ras signaling to the prevention of p53 acetylation is currently unknown and requires further investigation.
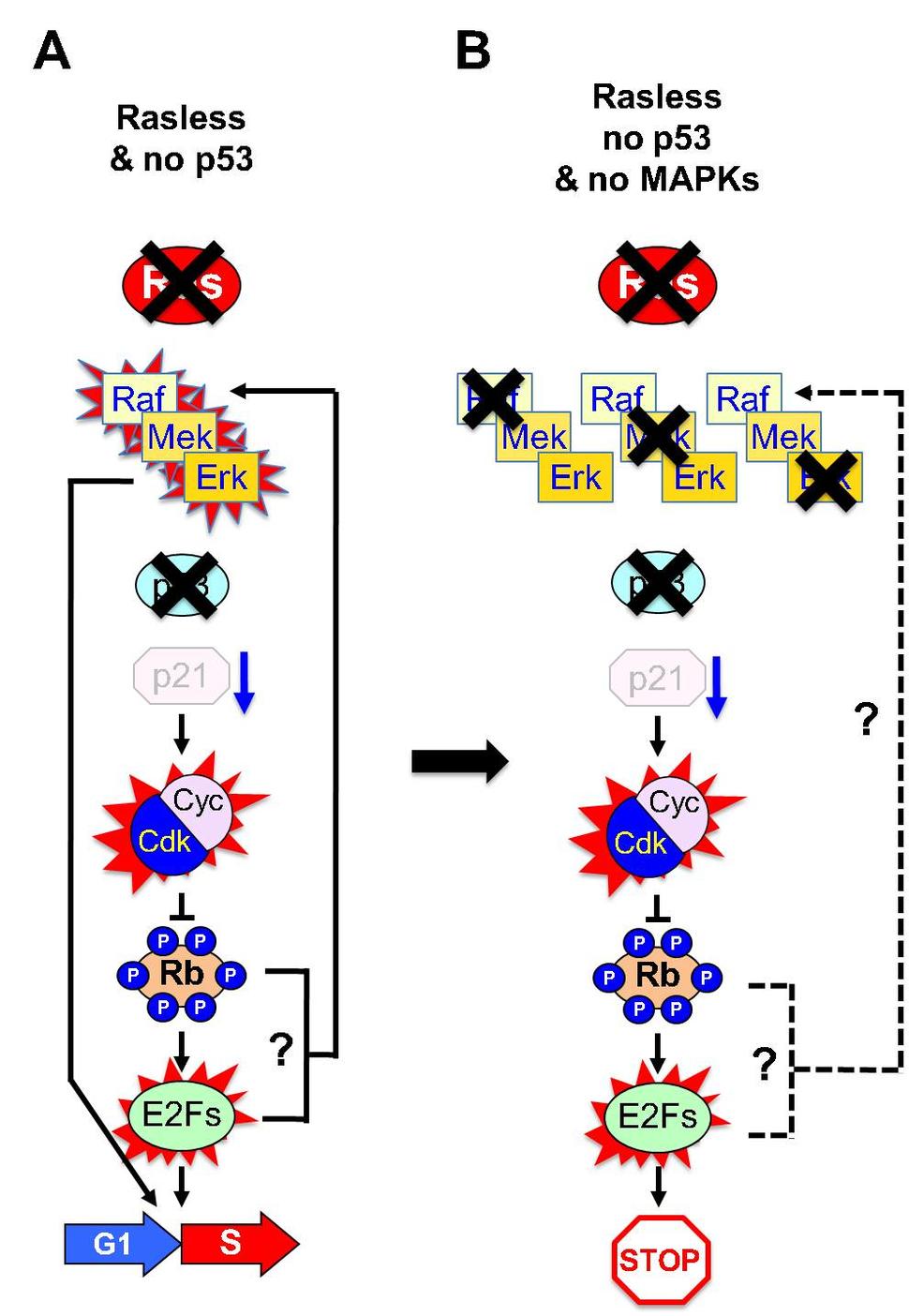
FIGURE 2: (A) Schematic representation of our understanding of how cells proliferate in the absence of Ras and p53. In the absence of both proteins, the level of inhibitory p21 drops, thus signaling through cyclin/Cdk, Rb, and E2Fs to stimulate cell cycling. Surprisingly, our data show activation of Raf, Mek, and Erk in the absence of any Ras proteins. The arrow from Rb and E2F indicates a possible connection between those proteins and Raf/Mek/Erk activation, however the mechanism is unknown. (B) Cell cycling in the absence of Ras and p53 requires the presence of Raf and Mek and Erk. Inhibition of any one of these abolishes cell cycling. The dotted line indicates that in Rafless, Mekless or Erkless cells we do not have experimental evidence for the retroactivating circuitry observed in Rasless cells. The represented active state of the E2F transcription factors is deduced from the inactive state of the Rb protein, but it has not been determined experimentally.
Previous studies have revealed that only active isoforms of the MAP kinase signaling cascade were able to induce proliferation of Rasless cells3,4. In contrast, ectopic expression of activated forms of PI3Kinase p110alpha or RalGDS failed to overcome the quiescent state of these cells. These observations indicate that the Raf/Mek/Erk pathway is solely responsible for mediating Ras mitogenic signaling at least in MEFs and keratinocytes3,4. Indeed, cells lacking either the three Raf kinases (Rafless cells), the two Mek kinases (Mekless cells) or the two Erk kinases (Erkless cells) also exit the cell cycle and remain in a quiescent state indistinguishable from that of Rasless cells. In addition, Erkless cells display an active p53/p21Cip axis similar to Rasless cells. Thus, we decided to extend our studies to these cells assuming that they will also be able to exit quiescence upon knock down of the p53/p21Cip1 axis. Unexpectedly, knock down of either p53 or p21Cip1 failed to convey proliferative properties to Rafless, Mekless or Erkless cells. Knock down or inactivation of the Rb tumor suppressor also failed to induce cell proliferation in those cells.
A solution to this apparent conundrum came when we observed that knock down of any of the members of the p53/p21Cip1/Rb tumor suppressor axis in Rasless cells resulted in activation of the Raf/Mek/Erk signaling pathway (Figure 2A). Thus, these cells possess a retro-activation circuitry that maintains an active MAP Kinase cascade in the absence of both Ras and the p53/p21Cip1/Rb tumor suppressor axis. Moreover, these kinases are also essential for cell proliferation of Rasless cells (Figure 2B). In other words, inactivation of Rb per se is not sufficient to license cells to cycle, they require an active Raf/Mek/Erk pathway. These results open a series of important questions whose resolution will allow us to fully understand the cellular circuitries responsible for handling mitogenic signals. For instance, how does knock down of the p53/p21/Rb axis activate the MAPK pathway when Ras is absent? Since Raf proteins are essential for cell proliferation in the absence of an active p53/p21/Rb axis, we have to assume that the retro-activation circuitry must necessarily go through this family of kinases. It is possible that cells may also have other circuitries to retro activate the Mek and Erk kinases. But if so, our genetic data indicate that either these circuitries are not sufficient to sustain cell proliferations or they will also mediate retro-activation of the Raf proteins8. In any case, and regardless of the precise nature of these circuits, our genetic studies clearly illustrate the activation of the MAPK signaling cascade in the absence of Ras proteins. Moreover, they also unambiguously demonstrate that whereas Ras proteins are dispensable for cell proliferation in the absence of an active p53/p21/Rb axis, the individual components of the MAPK kinase cascade, the Raf, Mek and Erk kinases are absolutely essential.
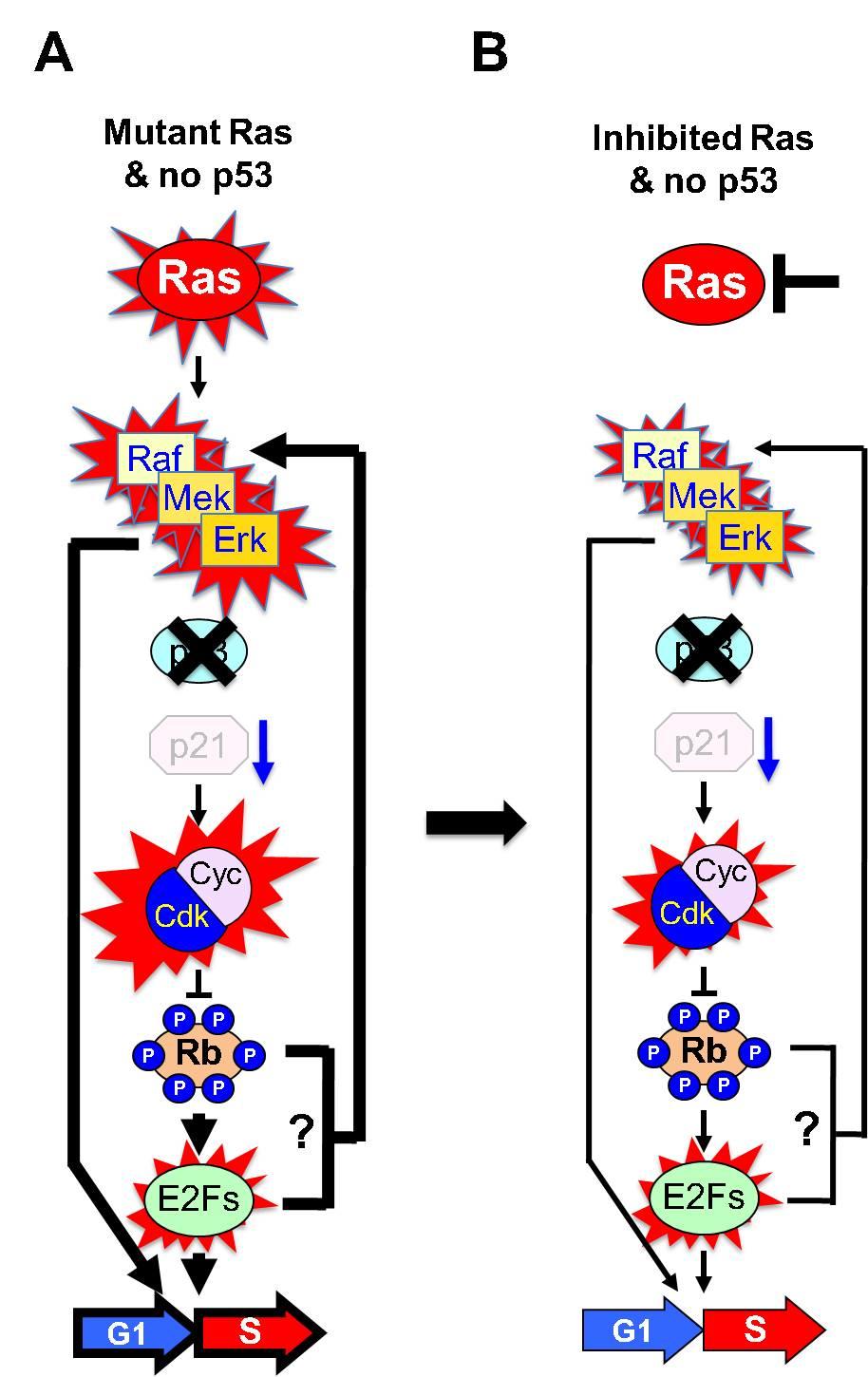
FIGURE 3: (A) Schematic representation of presumed cooperation between mutant Ras and loss of p53 in cancer. In this scenario, loss of p53 would reinforce activation of the MAPK pathway by mutant Ras, which in turn may cause stronger or more efficient MAPK activation, amplified signaling, and increased cell proliferation (indicated by bold arrows). (B) Our results suggest that even if an effective inhibitor of Ras signaling were to be developed (no arrow from Ras to Raf), the loss of p53 that is common in many tumors would result in constitutive activation of Raf, Mek, and Erk, although probably at lower levels compared to uninhibited oncogenic Ras (indicated by thinner arrows). Such cancer cells might retain tumorigenic characteristics even if signaling from oncogenic Ras is absent.
Our observations raise the possibility that the feedback activation of the Raf/Mek/Erk pathway downstream of the p53/p21/Rb axis may also exist during normal homeostasis as a mechanism to potentiate mitogenic signaling9. Yet we know that cells with normal Ras proteins but lacking an active p53/p21Cip1/Rb axis do not proliferate without signaling through Ras, indicating that the intensity of MAPK pathway activation through p53/p21Cip1/Rb loss may not be sufficient to elicit unscheduled cell cycle entry in most cells. Alternatively, cells may have negative regulatory mechanisms that restore activation of the p53/p21Cip1 axis once they stop receiving mitogenic signals. Nevertheless, it is tempting to speculate that such a retroactivated MAPK pathway may contribute to a more sustained or durable activation of Erk kinases, which, in contrast to transient activation, has been shown to be promote efficient entry into the cell cycle10.
A final consideration derived from our genetic studies is whether cells in human tumors lacking the p53 tumor suppressor may retain an active MAPK signaling pathway independent of Ras signaling. If so, direct inhibition of Ras proteins may not be sufficient to stop tumor progression11 (Figure 3). Therefore, the identification of such a Ras-independent MAPK activation mechanism should provide vital information as to how tumors may escape direct Ras inhibition. In any case, it may also simply be possible that activation of Ras by point mutations and loss of p53 cooperate in tumors to potentiate MAPK signaling via Ras-dependent and -independent mechanisms. This hypothesis would provide an alternative explanation for the increased tumor progression rates in cancers that harbor a Ras oncogene as well as an inactive p53/p21Cip1/Rb axis12. Hence, our work raises the possibility that preventing the retro activation of the MAPK signaling pathway adds a novel aspect to the role of p53 as a tumor suppressor (Figure 3).
References
- Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer 2003; 3:459-65.
- Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 2011; 11: 761-74.
- Drosten M, Dhawahir A, Sum EY Urosevic J, Lechuga CG, Esteban LM, Castellano E, Guerra C, Santos E, Barbacid M. Genetic analysis of Ras signalling pathways in cell proliferation, migration, and survival. EMBO J 2010; 29: 1091-104.
- Drosten M, Lechuga CG, Barbacid M. Ras signaling is essential for skin development. Oncogene 2014; 33:2857-65.
- Peeper DS, Upton TM, Ladha MH, Neuman E, Zalvide J, Bernards R, DeCaprio JA, Ewen ME. Ras signalling linked to the cell-cycle machinery by the retinoblastoma protein. Nature 1997; 386: 177-81.
- Coleman ML, Marshall CJ, Olson MF. RAS and RHO GTPases in G1-phase cell cycle regulation. Nat Rev Mol Cell Biol 2004; 5: 355-66.
- Drosten M, Sum EY, Lechuga CG, Simón-Carrasco L, Jacob HK, García- Medina R, Huang S, Beijersbergen R, Bernards R, Barbacid M. Loss of p53 induces cell proliferation via Ras-independent activation of the Raf/Mek/Erk signaling pathway. Proc Natl Acad Sci USA 2014; 111: 15155-60.
- Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJS, Kornev AP, Taylor SS, Shaw AS. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 2013; 154: 1036-1046.
- Korotayev K, Chaussepied M, Ginsberg D. ERK activation is regulated by E2F1 and is essential for E2F1-induced S phase entry. Cell Signal 2008; 20: 1221-6.
- Meloche S, Pouysségur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007; 26: 3227-3239.
- Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013; 503: 548-51.
- Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, Jacks T. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res 2005; 65: 10280-10288.