The K-Ras Anchor: Another Twist to the Tale
, by John Hancock

John F. Hancock, M.B, B.Chir, Ph.D.
John Hancock received his medical training at the University of Cambridge and earned his Ph.D. from the Institute for Cancer Research, London. He moved to the US in 1992 where he was a research director at Onyx Pharmaceuticals, and then to Australia where he played a prominent role in the Institute for Molecular Biosciences, University of Queensland. He is now Chair of the Department of Integrative Biology and Pharmacology, and Executive Director of the Institute of Molecular Medicine, University of Texas Health Sciences Center at Houston. Hancock has published more than 100 papers on Ras proteins with an emphasis on their interactions with membranes.
Way back in 1990 we thought we had pretty much figured out how Ras proteins were anchored to the plasma membrane (PM). Ras anchors comprise two components, a triplet of posttranslational modification of the C-terminal CAAX box generates a farnesylated methyl-esterified cysteine, which then operates in conjunction with a second motif, which is either palmitoylation in the case of H-Ras and N-Ras or a polybasic domain (PBD comprising a sequence of 6 contiguous lysine residues) in K-Ras4B (hereafter K-Ras). PBDs are commonly found in the PM anchors of other small GTPases where they also operate in conjunction with a prenyl group (C15-farnesyl or C20-geranylgeranyl); the charged residues in these PBDs include lysines and arginines. Small GTPase PBD primary sequences are in fact all different. Nevertheless, PBDs are presumed to interact with PM exclusively via electrostatics such that their only relevant feature is the number of basic residues that will determine the strength of electrostatic association with generic anionic lipids in the PM.
Unlike other Ras proteins, K-Ras4B lacks palmitoylation sites in its C-terminal membrane-anchoring peptide. Instead its polybasic tail interacts with the anionic head groups of phosphatidyl lipids on the inner plasma membrane. Together with the lipid modifications at the C-terminus, the polybasic region of K-Ras4B displays and influences specific membrane interactions that affect how this potent oncogene signals to the rest of the cell. Reprinted from Cell 168, 239, 2017 with permission purchased from Elsevier.
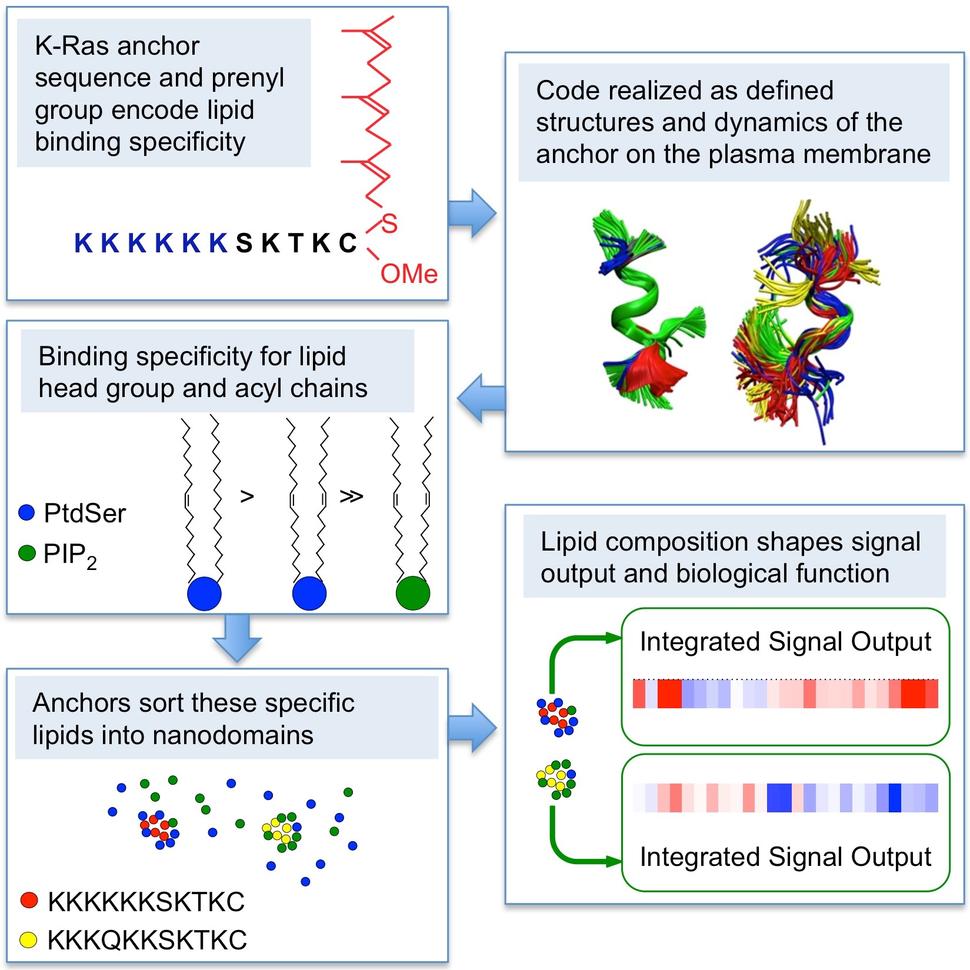
Our recent work using electron microscopy for spatial mapping of a diverse set of lipid probes, coupled with FLIM-FRET [fluorescence lifetime-fluorescence resonance energy transfer] imaging and lipid add-back experiments in intact cells, now shows that the K-Ras anchor is much more than a simple charge detector. Rather, the anchor actually encodes exquisite specificity for phosphatidylserine (PtdSer) over other anionic phospholipids, including phosphatidylinositol (4,5) bis-phosphate (PIP2). This binding specificity also extends to PtdSer with very specific combinations of lipid chains. In this context several data show that lipid interactions of the K-Ras anchor are not simply governed by charge. Replacing the six lysines (6K) in the PBD with six arginines (6R), which have the same net positive charge, resulted in K-Ras-6R being assembled into nanoclusters with a very different lipid composition than K-Ras with a wild-type (6K) PBD. Switching the prenyl group from farnesyl to geranylgeranyl also changed the lipid composition of the 6K and 6R nanoclusters, indicative in each case of different lipid binding specificities of the intact anchors. In addition K-RasG12V with six different single point mutations of K→Q that scan across the PBD each assemble into nanoclusters with markedly different lipid compositions even though each PBD point mutant has the same net charge. Indeed the two point mutants K-Ras-K177Q and K178Q showed enhanced interaction with multivalent PIP2 concomitant with a loss of binding to PtdSer, thus a single point mutation in the PBD can switch lipid specificity from PtdSer to PIP2. Together these and other data show that the precise amino acid sequence of the PBD and prenyl group define a combinatorial code for lipid binding that extends beyond simple electrostatics; within this code lysine and arginine residues are non-equivalent and prenyl chain length modifies nascent PBD lipid preferences.
We next used extended (8 μs) classical molecular dynamics (MD) simulations and free energy calculations from metaMD of the K-Ras anchor on a 20% PtdSer : 80% phosphatidylcholine bilayer to solve the anchor structure. These experiments showed that the lipid binding code is realized by distinct dynamic tertiary structures of the anchor that govern amino acid side-chain-lipid interactions. For example the K-Ras 6K anchor adopts a combination of disordered, intermediate and ordered helical structures that present different lysines for interaction with the bilayer. Not all lysines of the PBD therefore participate equally in hydrogen bonding with the bilayer, but the side chains of K177, K178 and K180 are especially important. Whereas the K-Ras 6K anchor favors a disordered extended state and associates with PtdSer extensively, the single point mutations K177Q or K178Q shift the conformational equilibrium to more ordered states and compromise hydrogen bonding with PtdSer. Quantification of the different anchor variants also showed that PtdSer clustering around the anchor is not a simple function of electrostatic interactions: notably replacing C15-farnesyl with C20-geranylgeranyl, a substitution that does not affect charge distribution, nearly abolishes PtdSer clustering around the anchor. The farnesyl to geranylgeranyl switch induces markedly different conformational dynamics of the anchor peptide because the longer prenyl-chain length increases van der Waals interactions with bilayer lipids whilst concurrently decreasing hydrogen bonding with PtdSer head groups at the membrane interface. This result recapitulated data from EM spatial mapping experiments in intact cells which showed that geranylgeranylated K-RasG12V exhibits much less selectivity for different anionic lipids than does farnesylated K-RasG12V, perhaps reflecting the reduced head group interactions evident in the anchor structures.
In sum K-Ras interactions with membrane lipids are not simply governed by electrostatics: The precise sequence of the PBD and prenyl group together determine the conformational dynamics of the anchor, the residues participating in membrane binding and hence which anionic lipid interactions are favored. Subtle changes in PBD sequence, even single K→Q or K→R substitutions, as well as phosphorylation therefore have substantial effects on structural dynamics and hence lipid interactions. Interestingly phosphorylation at Ser181 rendered the predominantly disordered conformation of the anchor more peripheral than the non-phosphorylated anchor despite having additional lipid interactions via the phosphate group forming a novel hydrogen bond with the NH3 moiety of a PtdSer head group. In intact cells this change in anchor structure also switched lipid preference from PtdSer to PIP2. Previous work has shown that Ras G-domain also participates in PM binding, engaging differently in GTP-bound and GDP-bound conformations. Concordantly whereas the lipid preferences of GTP-bound K-RasG12V and the minimal K-Ras membrane anchor are similar, those of GDP-bound K-Ras are quite different, consistent both with the G-domain modulating lipid interactions of the anchor with the PM, and with the lateral segregation of K-Ras.GTP and K-Ras.GDP into different nanoclusters.
In the complex environment of the PM we also observed that the K-RasG12V anchor exhibits remarkable specificity for distinct subclasses of PtdSer, preferentially assembling asymmetric PtdSer with one saturated and one desaturated lipid chain into nanoclusters. K-RasG12V does not interact with fully saturated PtdSer at all, whereas mono- and di-unsaturated PtdSer can support K-RasG12V PM binding but cannot be assembled into nanoclusters. It seems most probable that these differences reflect the lateral availability of PtdSer in the PM. Fully saturated PtdSer is likely retained in cholesterol-rich complexes more extensively than asymmetric PtdSer, concordant with previous demonstrations of a cholesterol sensitive pool of PtdSer that does not interact with K-RasG12V. Moreover, membranes composed of asymmetric PtdSer sustain less perturbation when undergoing electrostatic interactions with external components than fully saturated PtdSer. These biophysical properties may account for the preferential sorting of asymmetric PtdSer into K-RasG12V nanoclusters. These results also illustrate the functional importance of nanoclusters, since only asymmetric PtdSer was able to fully restore K-RasG12V interactions with CRAF in PtdSer-depleted cells. Most importantly the distinct lipid binding specificity of each K-RasG12V PBD mutant translated into different extents of PM localization, nanoclustering and assembly of nanoclusters with different lipid compositions. The integrated effect of these differences was a unique signaling profile for each mutant, indicating a critical role for anchor lipid binding specificity in effector recruitment and activation and hence K-RasG12V signal output.
These findings have broader implications in understanding the function of the lipid-binding domains of other PM-binding proteins. Certain lipid-binding domains (e.g. C2 and PH/PX domains) have long been known to possess defined structures and bind to specific lipids with high affinity. By contrast, as already mentioned, many small GTPases have membrane anchors analogous to K-Ras comprising a PBD of lysine and arginine residues operating in conjunction with a prenyl lipid. Our results suggest that each of these anchors by virtue of their different primary sequences will exhibit a distinct secondary structure on the PM and hence have the capacity to selectively interact with and sort different cohorts of anionic lipids with attendant consequences for function, in turn promising many more twists in the tales of small GTPase membrane interactions and their anchoring mechanisms.