RAS and MYC: Co-conspirators in Cancer
, by Brittany L. Allen-Petersen and Rosalie C. Sears
Aberrant activation of RAS influences a variety of cellular pathways, the majority of which have been highlighted in RAS pathway v2.0. Despite this knowledge, efforts to attenuate RAS activity through the therapeutic inhibition of downstream signaling molecules have been relatively unsuccessful, in large part due to the ability of RAS signaling pathways to “rewire”, increasing therapeutic resistance1,2. Given our current inability to directly inhibit RAS, identification of an essential downstream target in RAS-driven cancers would be highly valuable. Here, we discuss RAS-mediated posttranslational modifications (PTMs) of c-MYC (MYC) that increase MYC activity and position MYC as a critical effector of RAS-driven cancer.
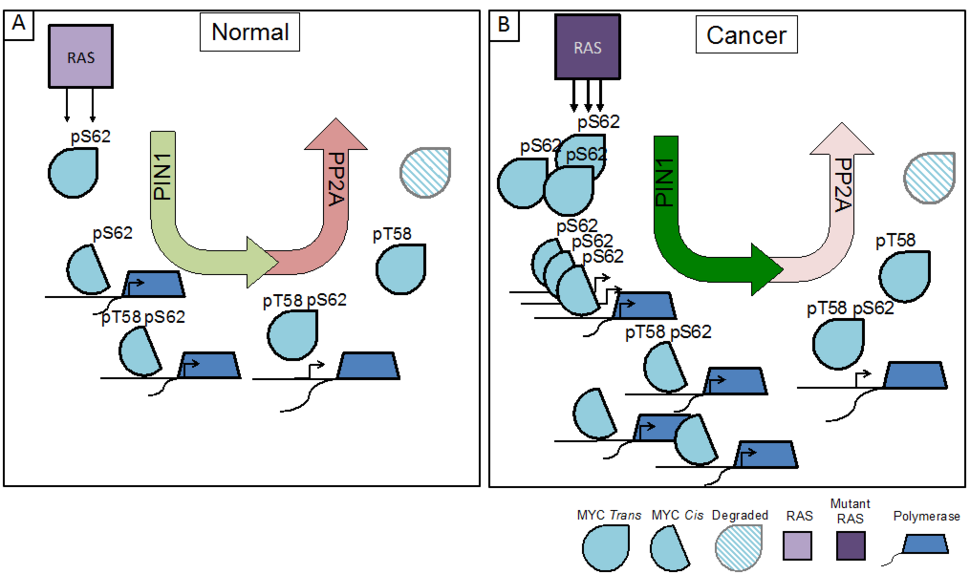
A. In normal cells, RAS signaling leads to MYC phosphorylation at S62, which supports PIN1 isomerization of P63 to cis, recruitment of MYC to target gene promoters, and activation of the basal transcription machinery. Subsequent phosphorylation of MYC at T58 results in a second PIN1 isomerization of P63 to trans that is associated with the release of MYC from DNA, PP2A mediated pS62 dephosphorylation and MYC degradation.
B. In RAS-driven cancer, increased signaling from mutant RAS along with active PIN1 and suppressed PP2A leads to an accumulation of active MYC that can drive pro-tumor transcriptional programs.
MYC is an oncogenic transcription factor that regulates a broad set of gene programs essential to growth, differentiation, and proliferation3. Given the almost ubiquitous nature of MYC genomic targets, MYC expression is tightly regulated at the transcriptional, translational, and posttranslational level. While MYC gene amplification is considered to be an important driver of oncogenesis, MYC is also known to undergo posttranslational modifications (PTMs) that influence its location, stability, and activity, including acetylation, ubiquitinylation, SUMOylation, glycosylation, phosphorylation, and phosphorylation-directed isomerization4-6.
For example, studies have defined a posttranslational phosphorylation cascade downstream of RAS that significantly impacts MYC activity. Under normal growth conditions, RAS activation of MAPK and/or CDK leads to the phosphorylation of MYC at Serine 62 (pS62-MYC). This form of MYC is recognized by the phosphorylation-directed peptidyl-prolyl isomerase, PIN1, which isomerizes Proline 63 (P63) from trans to cis, enhancing MYC’s recruitment to DNA and leading to increased expression of MYC target genes. Upon relief of RAS-PI3K-AKT suppression of GSK37, phosphorylation of S62 also primes MYC for GSK3-mediated phosphorylation at Threonine 58 (pT58-MYC). The pS62/pT58 form of MYC undergoes a second proline isomerization at P63 (cis to trans) by PIN1, thereby licensing the protein phosphatase PP2A to return MYC to its dephosphorylated state at S62. This form of MYC is released from DNA and degraded through ubiquitination and proteolysis by the 26S proteasome (Figure 1A)8-14. Together, this phosphorylation/isomerization cascade results in regulated interaction of MYC with target gene promoters, where the dynamic binding and release of MYC increases gene expression and allows for gene re-activation and/or gene network rewiring in response to changes in MYC availability9,15.
In cancer, the pathways that lead to the phosphorylation of MYC at S62, such as those driven by oncogenic RAS, are often upregulated, and pathways that lead to the degradation of MYC are frequently suppressed, resulting in a high overall pS62/pT58-MYC ratio16,17. This shift in phosphorylation provides a stabilized pool of MYC that can readily bind promoters and enhancers to drive proliferative and neoplastic gene programs (Figure 1B)9,18. In addition to the increased phosphorylation of MYC, RAS has also been shown to increase PIN1 expression19,20 and suppress PP2A activation21, highlighting the importance of MYC deregulation to RAS oncogenic signaling programs.
Historically, RAS and MYC are the quintessential example of cooperating oncogenes22. Recent studies have demonstrated a critical effector role for MYC: loss of one copy of Myc in a KrasG12D-driven mouse (KPC) model of pancreatic cancer significantly prolongs survival and restricts lesions to a benign precursor stage23 and the genetic expression of a dominant negative form of MYC, OmoMyc, results in tumor regression in KRAS-driven lung adenocarcinoma, as well as stromal normalization24,25. While high expression of Myc can drive tumorigenesis in several tissue types, mice expressing transcriptionally deregulated, but physiological levels of Myc under the ROSA promoter exhibit normal histology26. However, when combined with mutant Kras, this low-level, deregulated Myc significantly accelerates the formation of pancreatic lesions in vivo as compared to KRAS expression alone (our unpublished results). These studies highlight the importance of deregulated MYC expression in KRAS driven transformation.
Similar cooperation has been seen in tumor models that combine mutant KRAS with altered expression of proteins that post-translationally control MYC activity. Loss of Pin1 results in an overall resistance to oncogenic drivers, including RAS-driven mammary tumorigenesis in vivo27. Similarly, loss of Fbw7, the E3 ubiquitin ligase that targets MYC for proteosomal degradation, results in accelerated KrasG12D-driven pancreatic tumorigenesis28. Consistent with these results, preliminary data in our lab suggests that decreased expression of PP2A B56α, the subunit attributed to dephosphorylating pS62-MYC29, increases acinar cell plasticity and pancreatic lesion formation in combination with mutant KRAS in vivo. By taking into account the altered expression and/or activity of enzymes that post-translationally modify MYC, post-translational deregulation of MYC is likely a common occurrence in cancer, and as the above genetic studies indicate, this appears particularly relevant to RAS-driven cancers. In this regard, it is notable that a recent study by the Der lab exploring ERK inhibition in KRAS-driven pancreas cancer identified the post-translational stability of MYC as an indicator of sensitivity or resistance to ERK inhibition, suggesting that therapeutic inhibition of MYC may be required for clinical efficacy30.
Given the central role of MYC as a mediator of multiple cell proliferation and survival pathways, it has been assumed that inhibition of MYC may have detrimental effects on normal cells. However, mouse studies utilizing OmoMyc have demonstrated that periodic loss of MYC results in no observable toxicity25. Similarly to RAS, MYC lacks druggable structural features that makes direct therapeutic targeting difficult. As such, therapeutic compounds that target the pathways that control MYC expression and/or drive the posttranslational regulation of MYC have emerged as novel cancer therapeutics. For example, BRD4 inhibitors have gained wide attention for their ability to decrease MYC expression31,32.
Other anti-MYC strategies aim to increase the phosphatase and inhibit the proline isomerase activities of PP2A and PIN1, respectively. PP2A has been shown to be a critical tumor suppressor in multiple tissue types33,34. Due to the large number of cellular PP2A targets, PP2A activity is controlled by multiple mechanisms including target-specific subunits, PTMs, and interaction with cellular inhibitors. Over the last decade, researchers have developed several compounds that increase PP2A activity, primarily by disrupting the interaction of PP2A with its inhibitor, SET33,35,36. Recently, Sangodkar et. al. demonstrated the therapeutic efficacy of SMAPs, first-in-class small molecule activators of PP2A, which are orally bioavailable compounds that activate PP2A by inducing a conformational change that alleviates cellular inhibition37. Treatment of mutant KRAS lung cancer cells with these SMAPs reduced oncogenic signaling and tumor growth in vivo. Using these compounds, our lab has generated similar results in KrasG12D-driven pancreatic cancer mouse models, with SMAP treatment attenuating the pathways most commonly attributed to therapeutic resistance including AKT, mTOR, ERK, and MYC, and extending survival. Despite having profound effects on multiple signaling pathways, SMAPs show no observable toxicities at high doses in vivo37. The absence of adverse side effects supports reports indicating that PP2A inhibition is a common occurrence in cancer cells, while normal cells require functional PP2A activity for the rapid on/off of proliferative and survival pathways and are, therefore, relatively unaffected by inhibiting cellular inactivation of PP2A35,38-42.
The proline isomerization mediated by PIN1 is thought to predominately promote cancer processes, as it is overexpressed in many cancers and increases the stability and/or activity of several pro-tumorigenic factors, such as AKT, HER2 and MYC19. Despite the large number of cellular targets, PIN1 null mice are viable43. In in vivo models of cancer, PIN1 null mice are resistant to several oncogenic drivers, including mutant RAS; conversely, PIN1 overexpression can increase RAS-driven phenotypes, suggesting that PIN1 is an important mediator of RAS signaling27. Therapeutic inhibition of PIN1, with either the synthetic compound PiB or the natural product Juglone, has shown efficacy in multiple cancer types44. Unfortunately, most of the PIN1 inhibitors show off target effects or have reduced potency in vivo. Recently, Wei et. al. demonstrated that PIN1 is a direct and critical target of all-trans retinoic acid (ATRA)45. Treatment with ATRA reduced PIN1 levels and decreased the growth of both triple negative breast cancer and acute promyelocytic leukemia cells in vivo. These studies provide insight into how we can leverage PIN1 inhibition for cancer therapeutics and potentially improve upon current ATRA compounds for increased efficacy. While the authors did not extend these studies to look at MYC activity, work from our lab and others have shown that inhibition of PIN1 with PiB significantly reduces MYC promoter binding and target gene expression9.
Although the posttranslational regulation of MYC can significantly contribute to RAS-driven cancer and to resistance to interventions targeting RAS signaling, many questions still remain. What MYC PTMs are necessary for MYC/RAS cooperation, and how are they regulated? Does mutant RAS alter the localization and temporal regulation of MYC PTMs to provide a survival advantage? How can we use knowledge of RAS-regulated MYC PTMs to improve cancer therapeutics? An increased effort to understand MYC posttranslational regulation and to develop therapeutic strategies that target MYC activity through the enzymes that modify it could be an important direction for the RAS community.
Brittany Allen-Petersen, Ph.D. (left) is a postdoc studying the role of phosphatases in regulating KRAS signaling in cancer. Rosalie Sears, Ph.D. (right) is a Professor in the Department of Molecular and Medical Genetics and Co-Director of the Brenden-Colson Center for Pancreatic Care at Oregon Health and Science University. The Sears lab studies cellular pathways that control tumor cell phenotypes, with a focus on the mechanisms that regulate c-MYC expression and activity.