A Panel of Isogenic RAS-Dependent Cell Lines Developed at the Frederick National Laboratory
, by Rachel Bagni
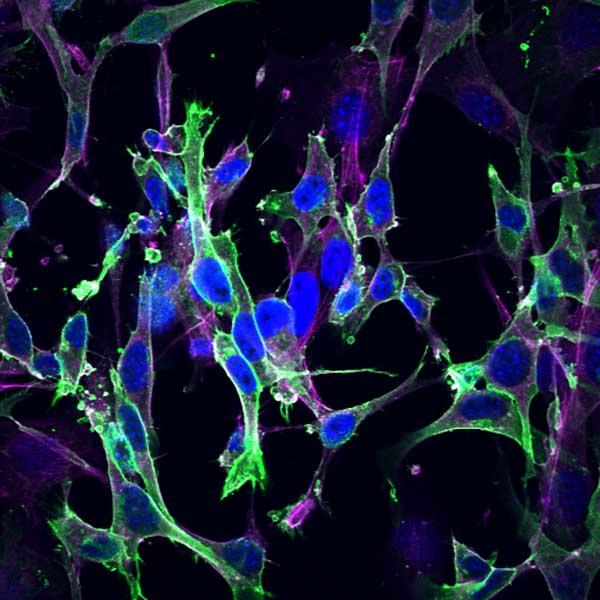
Mouse embryonic fibroblasts that were quiescent owing to deletion of all RAS genes were rescued with GFP-HRAS, and subsequently stained for nuclei (DAPI, blue) and actin (antibody, magenta).
A key undertaking of the NCI RAS Initiative is the development and validation of novel compounds that directly target the KRAS protein. The complex biology of RAS (see v2 of the RAS Pathway) and the clinical landscape of RAS driven tumors aside, we sought to develop a tool to clarify a question that consistently arises during early drug screening efforts: ‘Is my compound directly hitting (mutant) KRAS?’.
Building on the extensive work by Matthias Drosten, Mariano Barbacid and colleagues we used their RAS-less mouse embryonic fibroblast (MEF) cell line reagents to generate an isogenic and validated clonal panel of MAPK pathway-dependent cell lines (see Methods, below). The MEFs generously provided by the Barbacid laboratory are derived from NRAS- and HRAS-null mice and the KRAS gene has been floxed (removal by ER-Cre). Proliferation is dependent on the expression of either the endogenous KRAS gene or if it has been removed through tamoxifen treatment, the expressed transgene. Our panel includes the common clinical variants and an additional cell line dependent on the expression of BRAF V600E for proliferation.
These cell lines can be regarded as biochemical assays in a cellular context. To date, these rescued lines have been extremely useful in the RAS Initiative and for pharmaceutical and other external partners in screening for drugs that, for example, differentially inhibit proliferation of mutant KRAS over wild-type HRAS cells. For compounds that show this specificity, a secondary assay comparing mutant KRAS cells to mutant (V600E) BRAF cells provides data about whether the target is KRAS itself or another activity in the cell.
Rescue, proliferative, and genomic characteristics of the lines in the panel are shown in the table. Inquiries regarding these cell lines should be addressed to William Burgan.
| NCI-Frederick Designation | Expressed Transgene | Integration Sites | Exome Sequenced | Loss of Endogenous KRAS verified | Trp53 Status | Doubling Time (hrs) |
|---|---|---|---|---|---|---|
| RPZ26187 | KRAS 4A WT | 1 | Yes | WB, Exome | WT | 17 |
| RPZ25854 | KRAS 4B WT | Multiple | Yes | WB, Exome | WT | 26 |
| RPZ26216 | KRAS 4B WT | Multiple | Yes | WB, Exome | WT | 33 |
| RPZ26186 | KRAS 4B G12C | 1 | Yes | WB, Exome | WT | 21 |
| RPZ26198 | KRAS 4B G12D | 1 | Yes | WB, Exome | WT | 23 |
| RPZ26425 | KRAS 4B G12V | 1 | Yes | WB, Exome | WT | 39 |
| RPZ26299 | KRAS 4B G13D | Multiple | Yes | WB, Exome | WT | 39 |
| RPZ26295 | KRAS 4B Q61R | 1 | Yes | WB, Exome | WT | 41 |
| RP200024 | HRAS WT | Multiple | Yes | WB, Exome | WT | 21 |
| RPZ26379 | NRAS WT | 1 | Yes | WB, Exome | WT | 36 |
| RPZ26275 | BRAF V600E | 1 | Yes | WB, Exome | WT | 26 |
|
||||||
Methods
Primary mouse embryonic fibroblasts expressing only KRAS (DU1473; Hras-/-; Nras-/-; Kraslox/lox; RERTert/ert) were generated and characterized previously (EMBO J 29, 1091, 2010; PubMed PMID: 20150892) and obtained from Dr. Mariano Barbacid (CNIO, Madrid, Spain). The cells were cultured in the presence of 600 nM 4-hydroxytamoxifen to activate translocation of the estrogen receptor (ER)-fused Cre to the nucleus for removal of the endogenous (floxed) Kras genes. After arresting in the G1 phase (11-14 days) the cells were transduced with lentivirus to express human genes, either RAS genes or constitutively active BRAF (BRAF V600E). Cell proliferation resumed and transduced cells were selected using either puromycin or blasticidin and expanded to generate cell line pools that were dependent on expression of the exogenous transgene for continuous proliferation.
Clonal cell lines were derived from the pools using limited dilution single cell cloning. Clonality was confirmed using digital droplet PCR assays to count the copies of the lentivirus primer binding sites and the copies of the transferrin receptor gene or the TERT gene (ratios expected to be in agreement). Clones with ratios near 1 (suggesting 1 copy of the integrated virus per cell) or greater than 1 (suggesting multiple copies) were then analyzed using an integration sequencing assay (J Virol 81, 6731, 2007; PubMed PMID: 17409138) which precisely mapped the integration site(s) of the 5’ and 3’LTRs of the integrated lentivirus by alignment to the mouse genomic sequence. Single integrations of some genes did not rescue efficiently enough for isolation of clones. For these genes clones with multiple integrations were cultured, sub-cloned, and analyzed again to confirm stable populations.
Additional characterization of clones included confirmation of endogenous Kras gene removal (by western blots and sequencing), calculation of proliferation rates and doubling times (over 5 days), analysis of signaling pathways, and response to tool compounds. The exomes of all the lines were sequenced to exclude lines with mutations in genes known to be relevant to oncogenic processes.