RAS Initiative Computational Chemistry Research
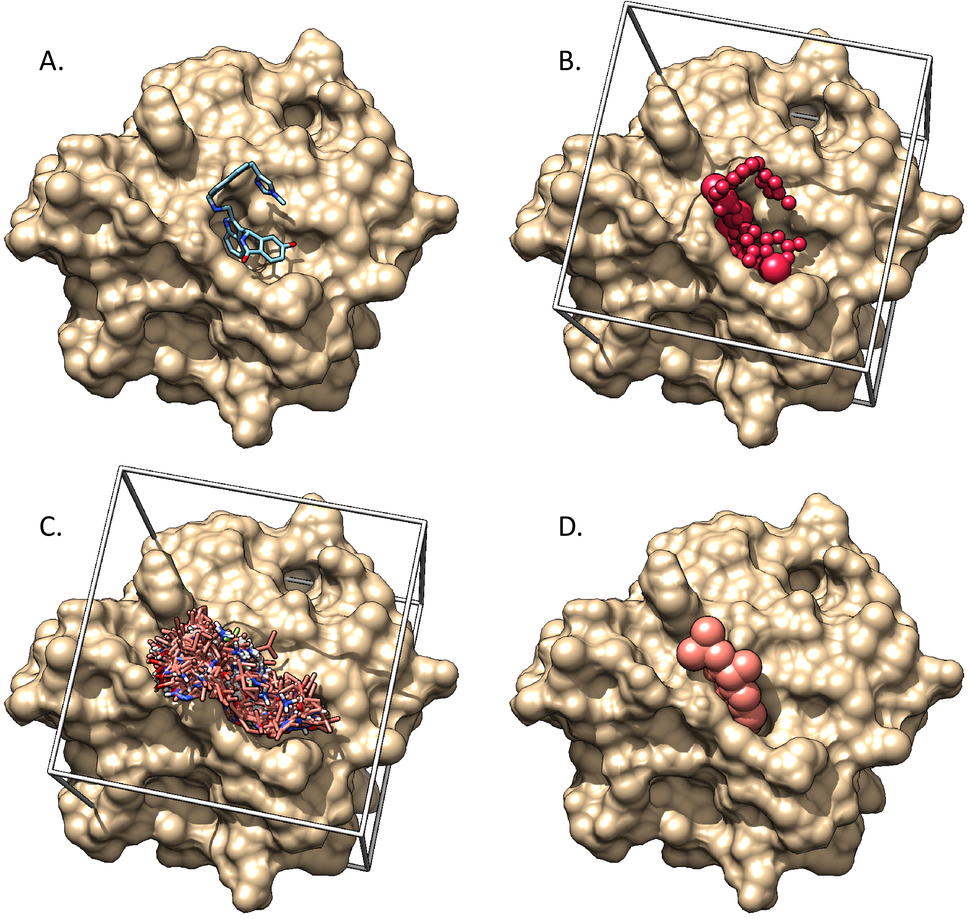
DOCKing to the BI-2852 pocket on KRAS. A. The ligand and receptor from the crystal structure. B. The pocket defined by spheres and grid box. C. Top 1,000 molecules from a screen of 23 million fragments. D. A top scoring molecule from the screen in space filling mode.
The RAS Initiative Computational Chemistry Research Team uses computers to aid in drug discovery. Our main tool is the software package UCSF DOCK, which we use for performing large-scale screens of hundreds of millions—soon to be billions—of molecules. To support our docking calculations, we also use chemical informatics methods, to depict, compare, and search for chemicals in a computer, and molecular dynamics simulations, to study the movement of atoms and molecules on a computer, combined with free energy calculations, to quantify the likelihood a chemical will interact with a receptor (protein) in a given configuration. We are highly collaborative and support wet lab teams within the RAS Initiative.
Progress
We have demonstrated that the small molecule BI-2852 stabilizes a RAS dimer. We also are contributing to many of the ligand discovery projects within the RAS Initiative. We are contributing to UCSF DOCK development by implementing new features and fixes, as well as participating in the DOCK community, a collection of several groups located around the world that use and/or develop DOCK and meets regularly.
Projects
- Ligand discovery against RAS and related targets.
- DOCK development
Tools
To aid in drug discovery efforts targeting RAS proteins, we use the following methods:
- Large-scale Molecular Docking of hundreds of millions of molecules for ligand (drug) discovery
- Molecular Dynamics Simulations to study small molecule binding and interactions
- Free Energy Calculations to quantify the quality of binding of small molecules
- Chemical Informatics for chemical comparisons and searches of the ZINC database
The RAS Initiative Computational Chemistry Research Team

Dr. Trent Balius, RAS Comp. Chem. Team Lead
Team lead and contact
Dr. Trent E. Balius, Ph.D.
trent.balius@nih.gov
301-846-6494
Collaborators
We are collaborating on developing new features into DOCK and applying them in discovery campaigns.
- Brian Shoichet, Ph.D., University of California, San Francisco
- Marcus Fischer, Ph.D., St. Jude Children's Research Hospital