Updates & Insights Blog
This blog features current topics in cancer genomics research and news and updates from OCG. Let's continue the conversation on our Personal Genomics Podcast.
At the present time, all NIH-sponsored meetings are cancelled. We apologize for any inconvenience this may cause and appreciate your understanding.

-

Organoids and other next-generation cancer models are a key technology in precision oncology research. CCG staff will be presenting at AACR 2023 on HCMI, NCI’s program generating hundreds of advanced cancer models for researchers.
-
By uncovering new cell states and how cell states may change, researchers are gaining much needed insights into how small cell lung cancer develops and progresses. Dr. Charles Rudin and Dr. Dana Pe’er from NCI’s Human Tumor Atlas Network (HTAN) discuss how they are using single-cell technologies to learn more about this deadly form of lung cancer.
-
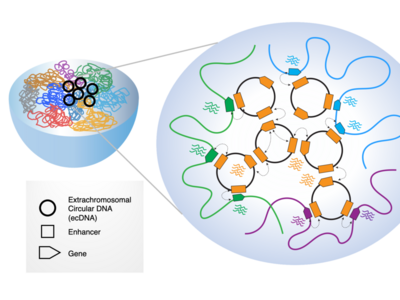
Extrachromosomal DNA (ecDNA) are emerging as oncogenic elements in cancer genomes. Dr. Chia-Lin Wei, Dr. Roel Verhaak, and Dr. Howard Chang are studying the interactions ecDNAs have with chromatin and amongst each other, revealing novel mechanisms of activating oncogenes and suggesting a bigger role for ecDNAs in cancer than previously appreciated.
-
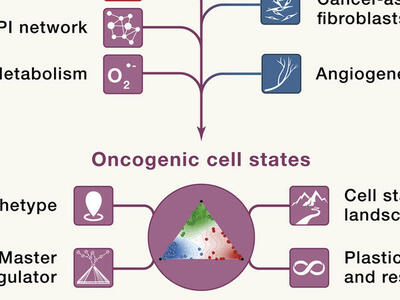
Identifying new targets and therapies for cancers will require thinking beyond the proverbial oncogene. Researchers from NCI’s Cancer Target Discovery and Development (CTD2) Network discuss new ways to model cancer and its potential vulnerabilities.
-

Identifying the “targets” for targeted therapy is a major challenge in precision medicine research. Dr. Jason Sheltzer discusses how he inadvertently uncovered mischaracterized cancer targets and what his findings may mean for cancer drug development.
-

The molecular mechanisms by which HPV infection leads to cancer, especially in low- and middle-income countries experiencing high incidence and mortality rates, have yet to be clearly delineated. A recent genomic, epigenomic, and transcriptomic study of cervical tumors from Ugandan patients looks at specific types of HPV and the genomic alterations they cause.
-
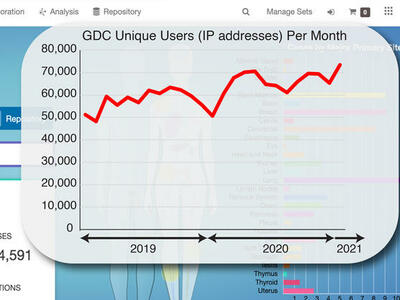
A short time ago the Genomic Data Commons (GDC) was just a dream. CCG Director Lou Staudt describes what the GDC team has built so far: an interactive data system with genomic data processing pipelines for all types of researchers to use.
-
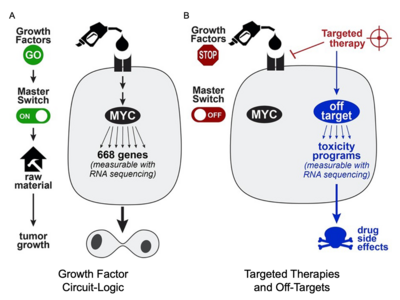
To better understand the off-target programs of cancer therapies, CTD² researchers created PANACEA, a gene expression database in response to 400 FDA-approved or late stage clinical cancer drugs, and hosted a community challenge to identify drug off-targets.
-

New features of HCMI's Searchable Catalog allows users to easily locate models of interest and their associated data at the Genomic Data Commons. HCMI is generating next-generation cancer models with quality-controlled clinical, biospecimen and molecular characterization data for the research community.
-

Recent updates to the CTD² Dashboard include changes to the presentation, annotation of experimental methods, search function, and addition of new links to external data resources and web applications. The CTD² Dashboard aims to make functional data and results from the CTD² Network easily accessible.