AT/RT is a primary central nervous system (CNS) tumor. This means it begins in the brain or spinal cord.
To get an accurate diagnosis, a piece of tumor tissue will be removed during surgery, if possible. A neuropathologist should then review the tumor tissue.
What Is the Grade of AT/RTs?
Primary CNS tumors are graded based on the tumor location, tumor type, extent of tumor spread, genetic findings, the patient’s age, and tumor remaining after surgery, if surgery is possible.
AT/RTs are all classified as grade 4 (also written as grade IV) tumors. This means they are malignant (cancerous) and fast-growing.
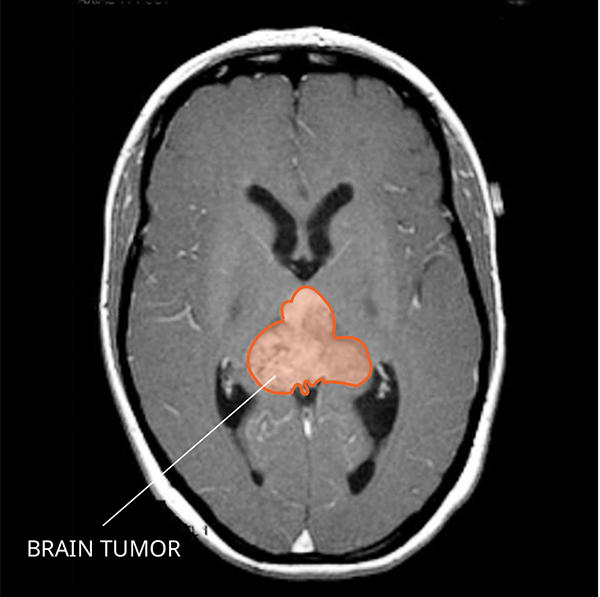
What Do AT/RTs Look like on an MRI?
AT/RTs usually appear very large with fluid-filled areas that often brighten with contrast on a magnetic resonance imaging (MRI) scan. You can often see areas of bleeding or dead tissue.
What Causes AT/RTs?
Cancer is a genetic disease—that is, it is caused by certain changes to genes that control the way our cells function. Genes may be mutated (changed) in many types of cancer, which can increase the growth and spread of cancer cells.
Most AT/RTs are caused by changes in a gene known as SMARCB1 (also called INI1), and less frequently by mutations in a gene called SMARCA4. SMARCB1 normally signals proteins to stop tumor growth. But, in AT/RTs, SMARCB1 doesn’t function properly and tumor growth is uncontrolled. In addition to occurring in the tumor’s DNA, SMARCB1 and SMARCA4 can also be found in a person’s own DNA. There are three groups of AT/RTs based on their genetic alterations: AT/RT-TYR, AT/RT-SHH, and AT/RT-MYC. Each group tends to develop in a different location of the CNS and is more common in different age groups. AT/RT-MYC is the most frequent group in adults.
Where Do AT/RTs Form?
AT/RTs can form anywhere in the CNS. They often occur in the brain and can spread to the spinal cord. AT/RTs in the brain can arise in the cerebral hemispheres, ventricles, suprasellar region, pineal gland, or the cerebellum and brainstem. AT/RTs can also begin in the spinal cord, but this is less common. They develop from several different very young cells called embryonal cells, including rhabdoid, neuroepithelial, epithelial, and mesenchymal cells.

Do AT/RTs Spread?
AT/RTs can be very fast-growing. They often spread to other areas of the CNS through cerebrospinal fluid (CSF). People diagnosed with AT/RT need tests (called staging) to determine if the tumor has spread to other areas. These tests include MRI examinations of the brain and spinal cord, as well as a spinal tap.
What Are the Symptoms of an AT/RT?
Symptoms related to an AT/RT depend on the tumor’s location and the person’s age. Here are some possible symptoms that can occur:
- Morning headaches
- Vomiting
- Changes in activity levels
- Loss of balance
- Increase in head size (in infants)
Because AT/RTs are fast-growing, symptoms usually get worse quickly.
Who Is Diagnosed with an AT/RT?
AT/RTs occur in both children and adults and are very rare in both age groups. Most patients are younger than two years of age at diagnosis. An estimated 470 people are living with this tumor in the United States and only 50 are adults. AT/RTs occur slightly more often in males than females.
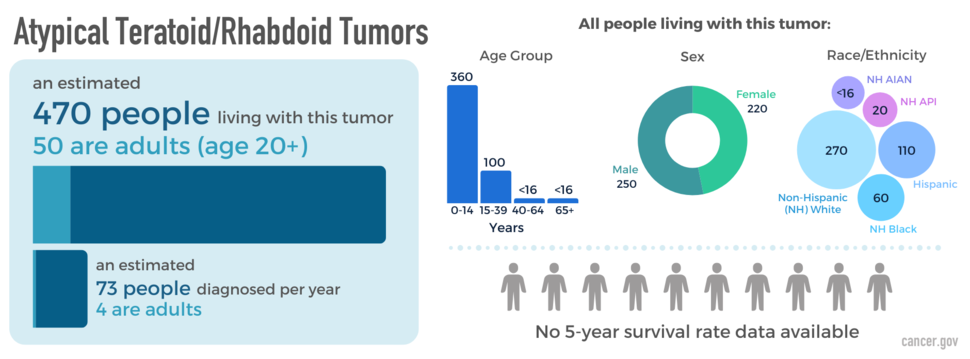
The blue box above shows the estimated number of people living with this disease (prevalence) as of December 31, 2019, as well as the average newly diagnosed cases per year (2008-2019) across all ages and adults (20+ years) specifically. The donut chart shows prevalence by sex; the bar graph shows prevalence by age group; and the packed circles chart shows prevalence by race and Hispanic ethnicity (American Indian/Alaska Native [AIAN]; Asian/Pacific Islander [API]). The people icons show the five-year relative survival rate in adults (2008-2018).
What Is the Prognosis of AT/RTs?
The likely outcome of the disease or chance of recovery is called prognosis. Prognosis is based on the tumor grade, location, tumor type, extent of tumor spread, genetic findings, patient’s age, and tumor remaining after surgery (if surgery is possible).
Many factors can affect prognosis, including the tumor grade and molecular type, the person’s age and health when diagnosed, and how they respond to treatment. If you want to understand your prognosis, talk to your doctor.
What Are the Treatment Options for AT/RTs?
The first treatment for an AT/RT is surgery, if possible. The goal of surgery is to obtain tissue to determine the tumor type and remove as much tumor as possible without causing more symptoms.
People with AT/RTs usually receive further treatments after surgery, which may include radiation, chemotherapy, or clinical trials. Clinical trials test new chemotherapy, targeted therapy, or immunotherapy drugs. Treatments are decided by the patient’s health care team based on the patient’s age, tumor remaining after surgery, tumor type, and tumor location.
Open Clinical Studies for AT/RTs
- PLX038 in CNS Tumors
- Immune Checkpoint Inhibitor Nivolumab for Patients with Rare CNS Cancers
- ONC206 for Patients with Rare CNS Neoplasms
Learn More
- Video: Clinical Trial Tests Nivolumab for Patients with Rare Brain and Spine Cancers
- Working Through a Life-Changing Rare Brain Tumor Diagnosis
- New Study Reveals Data on Rare CNS Tumors to Inform Clinical Care and Evaluate Novel Cancer Treatments
- Classifying Brain and Spine Tumors More Precisely to Improve Patient Outcomes and Care
- Read our NCI-CONNECTions Blog for recent news and information on rare brain and spine tumors.

Referrals
NCI-CONNECT doctors and nurses work with you and your primary doctor to collaborate on a comprehensive care plan that treats your brain or spine tumor. They will also help you cope with the physical and emotional aspects of your diagnosis. Learn about requesting a consultation >