New Treatment Target Identified for Key Prostate Cancer Driver
, by NCI Staff
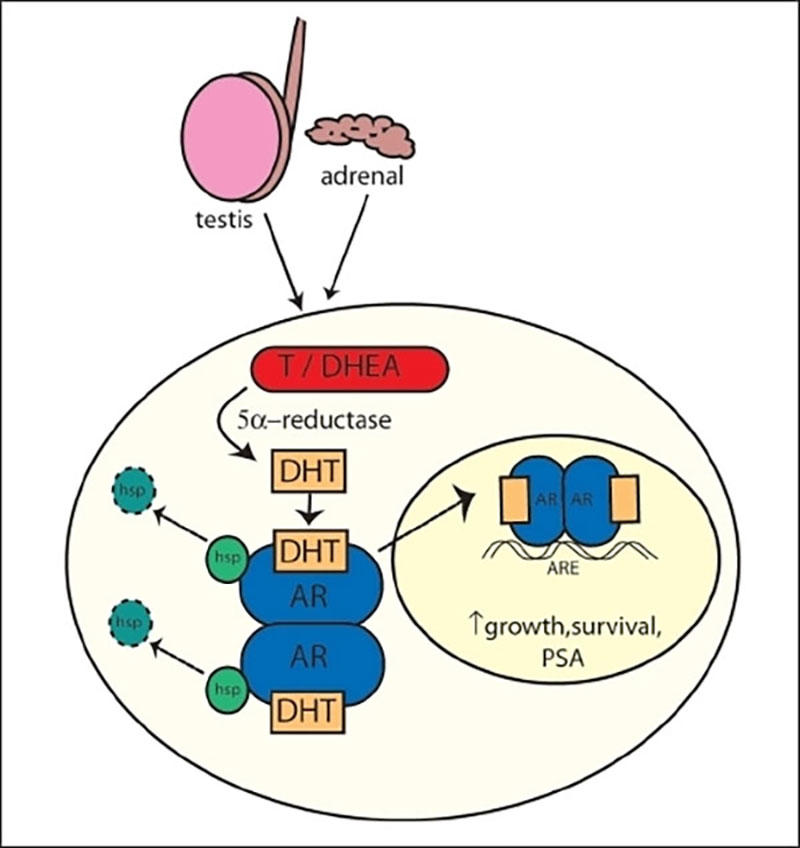
Androgen receptor (AR) in cells regulates the activity of several cancer-related genes. Signaling through AR is the chief means by which prostate cancer cells grow and spread.
Credit: Open-i (CC BY 2.0)
Researchers have identified a potential alternative approach to blocking the activity of a key molecular driver of an advanced form of prostate cancer, called androgen-independent or castration-resistant prostate cancer.
In prostate cancer cell lines and in several mouse models of prostate cancer, treatment with drug-like small molecules that target a protein called ROR-γ disrupted the activity of the androgen receptor (AR), the researchers showed. Signaling through AR is the chief means by which prostate cancer cells grow and spread.
In mouse models of castration-resistant disease, treatment with ROR-γ inhibitors led to substantial and prolonged shrinkage of tumors, and in mice with tumors that had been resistant to the AR-targeted therapy enzalutamide (Xtandi®), the treatment appeared to restore their sensitivity to the drug.
The study results were published March 28 in Nature Medicine.
Searching for an AR Regulator
Initial treatment of prostate cancer that has spread beyond the prostate mostly focuses on blocking androgens’ effect on tumors, typically by interfering with androgen production or blocking the action of androgens.
Eventually, however, most advanced prostate cancer becomes castration-resistant, continuing to progress and spread even when androgen levels are very low or undetectable, possibly because of changes to the AR protein or to its expression level, or due to mutations in the AR gene.
Over the last few years, several new drugs—including enzalutamide—have been approved for men with castration-resistant disease, but tumors almost invariably develop resistance to these therapies as well.
Because of AR’s pivotal role in the development of resistance to hormone therapies, much of the research on new therapeutic approaches in advanced prostate cancer has focused on this protein, said the study’s lead author, Hongwu Chen, Ph.D., of the University of California, Davis.
“One way or another, you have to address the androgen receptor,” he said.
To date, though, there has been little progress in directly targeting variant forms of AR or alterations in the AR gene that can promote treatment resistance. So Dr. Chen and his colleagues looked for another approach, searching for proteins that regulate the AR gene and, thus, affect AR synthesis.
By analyzing data from publicly available sources of tumor genomic data, the team homed in on a nuclear receptor protein called ROR-γ as a possible regulator of the AR gene. Nuclear receptors (which also include AR itself) are proteins that, when activated, bind DNA directly to regulate gene activity.
Following the ROR-γ Trail
Several lines of evidence pointed to the possibility that ROR-γ is involved in prostate cancer. The researchers found, for example, that the gene that codes for ROR-γ was overexpressed in metastatic castration-resistant tumors and that ROR-γ proteins were abundant in prostate tumors.
Further work in prostate cancer cell lines that overexpress AR suggested a direct relationship between ROR-γ and the receptor. Silencing the gene that codes for ROR-γ in prostate cancer cell lines, for example, decreased expression of the AR gene.
The researchers saw similar results when they treated cell lines with different ROR-γ inhibitors, including two drug-like molecules that are being developed to treat autoimmune disorders. The drugs had no effect on normal prostate cells or prostate cancer cell lines that don’t overexpress AR.
Additional experiments in cell lines showed that targeting ROR-γ affected the expression of cancer-related genes that are regulated by AR, including IGF1 and PTEN. And using the CRISPR/Cas9 system, the researchers identified how ROR-γ appears to regulate the AR gene, including identifying the likely DNA sequence where the protein binds to the gene and showing that deleting that section of the gene reduces its expression.
Finally, the research team tested the ROR-γ inhibitors in different prostate cancer mouse models, including models of prostate cancer that have resistance-related variants of the AR protein. The drugs halted tumor growth in all of the mouse models, except for a model of prostate cancer that did not overexpress AR.
In a mouse model of castration-resistant prostate cancer, the ROR-γ inhibitor SR2211 markedly reduced tumor size, but enzalutamide treatment did not. Combining the two drugs, however, led to more substantial and longer-lasting reductions in tumor size than the ROR-γ inhibitor alone, “which indicates that the ROR-γ antagonist can sensitize [castration-resistant prostate cancer] tumors to [enzalutamide],” they wrote.
“The idea of blocking the high expression of AR at the genetic level in these advanced cancers is not necessarily new,” explained Adam Sowalsky, Ph.D., head of the Prostate Cancer Genetics Section in NCI’s Center for Cancer Research.
But what’s exciting about this study, said Dr. Sowalsky, is the research team’s “use of new comprehensive datasets” to identify ROR-γ’s role as an actionable target in regulating expression of the AR gene. “This study certainly provides valuable proof-of-concept work to support further testing.”
Treatment with the ROR-γ inhibitors showed no evidence of toxic effects in the mice. “Some of the experiments were quite long, with treatment lasting as long as 50 days,” noted Dr. Chen, “and we still didn’t see any overt toxicity, like weight loss or organ dysfunction.”
The apparent lack of an effect on healthy tissue, the researchers wrote, may be due to “the addiction of tumor cells to elevated levels of AR, and to tumor cell-specific control of AR by ROR-γ.”
Despite the promising results, Dr. Sowalsky cautioned that there is still important work to do before drugs that target ROR-γ can be tested in men with castration-resistant prostate cancer.
In particular, he noted the need to test the drugs in more advanced models derived from human tumors that “harbor the complex drug-resistant phenotypes seen in today's prostate cancer patients.” These preclinical models, he noted, would “more reproducibly predict the actual response [in humans] to pharmacologic or genetic targeted therapies.”