3-D View of Mutations May Identify Potential Targets for Cancer Drugs
, by NCI Staff
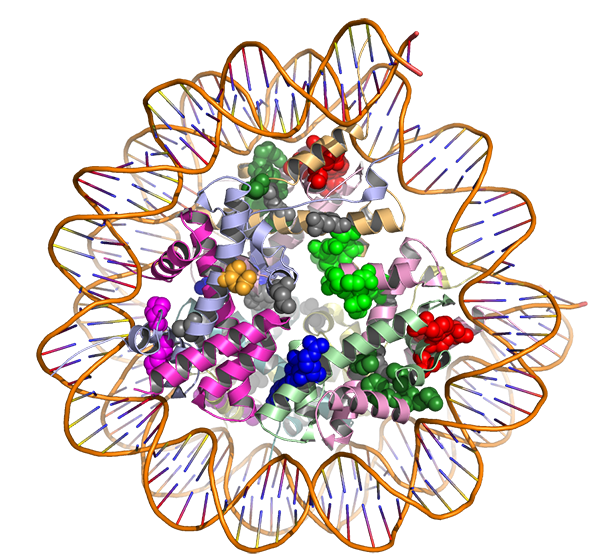
The HotSpot3D computational tool models how gene mutations (shown as bubble-shaped clusters) may affect the structure of proteins (spiral-shaped ribbons) as well as how those proteins may interact with a drug.
Credit: Washington University in St. Louis / Li Ding, Ph.D., and Feng Chen, Ph.D.
A new computational tool may help expand the known number of mutations in cancer cells that could be targeted with new or existing drugs.
Researchers recently reported that the tool, called HotSpot3D, allowed them to model how genetic mutations change the ways proteins function and interact with each other to potentially drive cancer. It also helped them identify more than 800 novel mutations that potentially could be targeted with existing drugs.
The research team that developed HotSpot3D, led by Li Ding, Ph.D., and Feng Chen, Ph.D., of Washington University in St. Louis, published results from this first large-scale testing of the computational tool June 13 in Nature Genetics.
2-D Instructions, 3-D Functions
DNA provides cells with a linear set of instructions for producing proteins. But when cells use those instructions to produce proteins, those proteins naturally fold into complicated 3-dimensional structures that dictate how they function.
Mutations in DNA that are linearly distant from each other (that is, far apart from each other on the DNA strand) may encode pieces of a protein that sit in close proximity to each other—and potentially affect how each functions—once that protein has folded into its active structure. Structural changes resulting from mutations can also alter how proteins interact with other proteins or small molecules that control their activity. For example, rendering a protein unable to bind to its normal cellular receptor or molecular regulators could cause faulty signaling in a molecular pathway, eventually leading to uncontrolled cell division and tumor development.
Tools that analyze some aspects of a genetic mutation's impact on protein structure have been available. But while sequencing glioblastoma samples for The Cancer Genome Atlas (TCGA), Drs. Ding and Chen realized the urgent need to develop more advanced tools for their research.
“We really wanted to understand how mutations interact with each other, and more importantly, we wanted to understand how mutations interact with drugs,” said Dr. Ding.
So, in a project almost 10 years in the making, the research team programmed HotSpot3D to map genetic mutation data from TCGA to two large, open resources that catalog protein structure. HotSpot3D uses these resources to model how genetic mutations might influence three different types of protein interactions: intramolecular (between pieces of a single protein), intermolecular (between two or more proteins), and drug–protein (between a protein and a drug designed to suppress or increase the activity of that protein).
“In sequencing data, you tend to have a vast amount of [DNA] variants that you discover in cancer samples," said Dr. Chen. "Some of them may have very little functional impact, but some of them may really be the drivers for cancers.” By teasing out which variants may actually change how proteins fold and work, HotSpot3D can help narrow down a subset of candidate mutations for further analyses, he explained.
Speeding Up the Sorting Process
In the Nature Genetics paper, the researchers applied HotSpot3D to more than 4,400 tumor samples sequenced by TCGA. These samples included about half a million mutations in 19 tumor types. The software identified more than 6,000 intramolecular and intermolecular interactions driven by mutations, some rare, that had potential significance to cancer development.
By comparing drug–protein interactions, HotSpot3D also identified more than 800 mutations that could potentially be targeted with existing therapies, such as drugs that inhibit a protein called BRAF. Many of the drugs that HotSpot3D predicted could bind to altered protein structures resulting from mutations are not traditional anticancer agents but other medications, including anti-inflammatory agents, calcium channel blockers, and anticonvulsants.
In proof-of-concept laboratory experiments, the researchers validated multiple mutations that HotSpot3D predicted would affect a protein called EGFR, a well-established driver of several types of cancer. In cell lines, several mutations were associated with higher-than-normal EGFR activity, even though they are located far from the EGFR gene in DNA.
Any results from HotSpot3D would need experimental validation, but the software potentially could help researchers rapidly sort through the large number of mutations found in any particular tumor and find an area of the genome to study further, commented Jacqueline Milne, Ph.D., head of the Electron Microscopy Core in NCI’s Center for Cancer Research.
This type of mutation–drug analysis could potentially be used to develop personalized treatment regimens, including regimens for diseases other than cancer, wrote the authors. They could also guide researchers interested in repurposing existing FDA-approved drugs, added Dr. Milne.
Targeted drugs will not exist for many of the mutations identified with HotSpot3D, but if clusters of mutations from different tumors all map to one three-dimensional space in a specific protein, drug developers might be very interested in looking closer at that area, concluded Dr. Milne.