DINO RNA Molecule Triggers Anticancer Response in Damaged Cells
, by NCI Staff
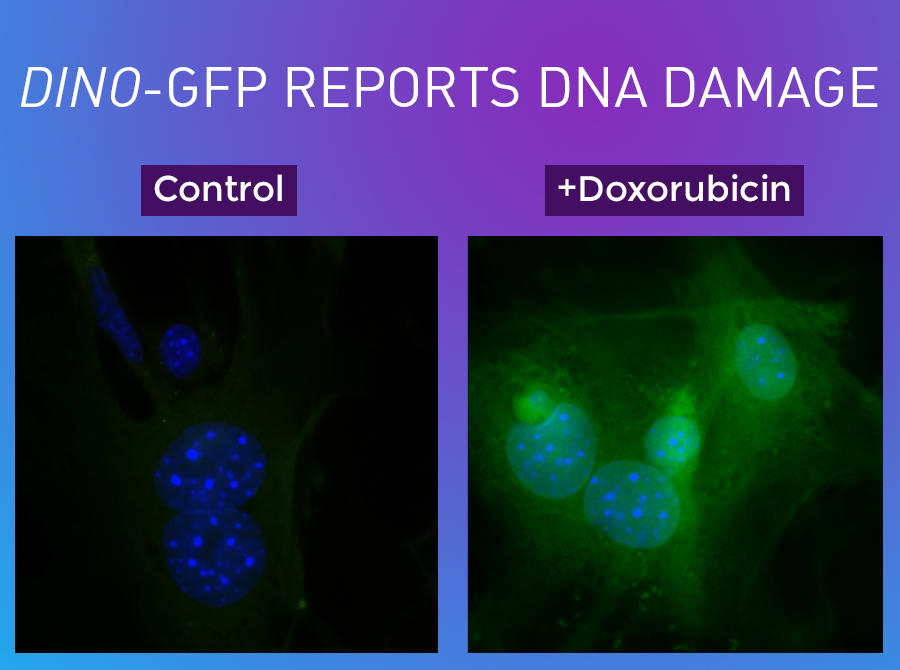
Left panel: Human fibroblast cells in their resting state. Right panel: After exposure to the DNA-damaging chemotherapy doxorubicin, the RNA DINO is produced in large quantities (green).
Credit: Julia Garcia, Stanford University
Researchers from Stanford University have identified an important molecule involved in the process that normally causes irreparably damaged cells to self-destruct. This molecule, a type of RNA they nicknamed DINO, helps regulate a vital tumor suppressor protein called p53, which is inactivated in more than half of all cancers.
In DINO, "we've found a new guardian of your genome," said Howard Chang, Ph.D., who led the study. His lab's experiments showed that, once cells accumulate a dangerous amount of genomic damage, the DINO (short for Damage Induced NOncoding) RNA takes part in a process that stabilizes the p53 protein so that it can initiate the self-destruct program, called apoptosis.
Cancer researchers would like to find ways to reactivate p53 and turn its tumor-suppressor function back on in cancer cells.
DINO—a previously unknown regulator of the DNA-damage response pathway driven by p53—could potentially be manipulated to achieve this reactivation, said Joanna Watson, Ph.D., of the Cancer Cell Biology Branch in NCI's Division of Cancer Biology, who was not involved in the study.
The study was published September 26 in Nature Genetics.
Flipping the Switch
Cells walk a fine line in terms of monitoring DNA damage and detecting when too much damage has occurred to remain viable. On one hand, they cannot be too quick to trigger the self-destruct process, because tiny mutations—most of which are harmless—accumulate every time a cell divides. On the other hand, if they wait too long, a cancer cell capable of uncontrolled growth can develop.
"You need a very sharp switch…so that when you get beyond the threshold of the cell's capacity for repair, you turn on the whole [self-destruct] program," explained Dr. Chang.
The p53 protein is known to play an integral role in this switch, but the molecular process that controls p53 itself has been unclear. "Normally, inside a cell, p53 is rapidly made and destroyed, so it's not really doing very much. It only becomes stabilized when there's DNA damage," said Dr. Chang.
"People knew that [p53 is] part of a surveillance mechanism, but how it switches from being unstable to being very stable and turning on a lot of genes to deal with DNA damage was not so clear. That's what this finding of DINO helps to resolve," he added.
Dr. Chang's team first noticed DINO while looking for DNA regions involved in cell division. DINO is a noncoding RNA, which means that although it is transcribed from DNA, it does not end up being translated into protein like other types of RNA. Instead, it performs functions within cells much like a protein would.
In the group's experiments, DINO turned out to have many interesting properties. When cells were exposed to high doses of the chemotherapy drug doxorubicin, which damages DNA, the expression of DINO jumped by 100-fold. If the researchers experimentally knocked out DINO before exposing cells to doxorubicin, many genes in the p53-controlled self-destruct signaling pathway were not turned on in response to the damage.
Other experiments showed that DINO physically interacts with the p53 protein, stabilizing it to prevent it from being broken back down within the cell.
Conversely, DINO could be used to "trick" cells into self-destructing. When the researchers used molecular techniques to add the RNA into cells that had not sustained DNA damage, the cells acted as if they had in fact been injured and triggered a damage response.
By adding DINO, the researchers are "forcing the cells into a [self-destruct] program that the cells actually weren't in. They're faking the cells out, for all intents and purposes," said Dr. Watson. This successful manipulation of the p53-driven self-destruct pathway "is one of the exciting elements of this paper," she added, as it hints at the potential to use DINO to switch on p53 on demand.
Harnessing DINO
In further experiments with mice, the researchers found that when mice lacking Dino (the mouse version of DINO) were exposed to what would normally be a lethal dose of radiation, they lived significantly longer than normal mice that expressed Dino, indicating that their cells were taking longer to "see" the damage they had accrued, though they did eventually die from radiation exposure.
The alteration of the damage response in this scenario is intriguing for its potential use in radiation therapy for cancer treatment, explained Dr. Chang. "This is something that you could [potentially] use to tune a patient's response to radiation, either to enhance radiation therapy or mitigate its side effects."
Further research is needed to explore these potential uses, as well as to investigate if manipulating DINO could actually prevent or reverse cancer cell formation resulting from a damaged p53 gene, added Dr. Chang.
His lab is planning follow-up studies to see how altering DINO expression affects cancer development and progression and to understand how DINO stabilizes p53.
While it might seem odd to call a molecule like DINO that triggers cell death a genome guardian, "it's a pruning process," concluded Dr. Chang. "If all these damaged cells stick around, and they're all still copying themselves, then we have a lot of problems."