Targeting the Accomplice: A Potential Treatment Approach for Rhabdomyosarcoma
, by Nadia Jaber
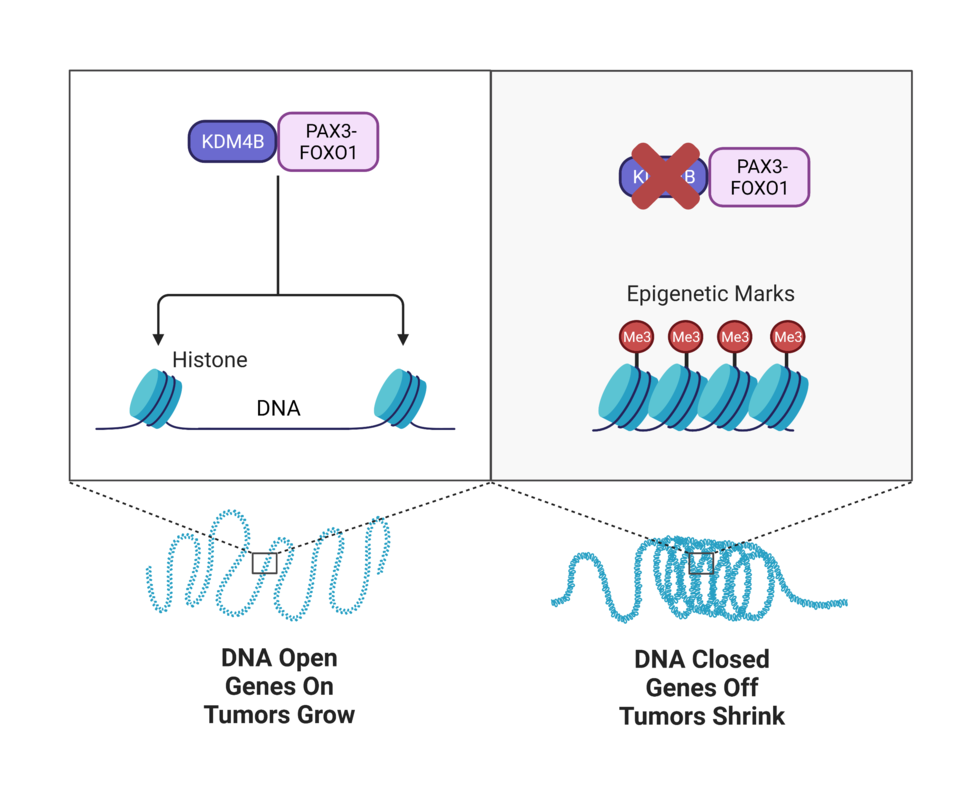
In mice with fusion-positive rhabdomyosarcoma, blocking KDM4B leads to epigenetic changes that close DNA, preventing the PAX3-FOXO1 fusion protein from turning on cancer-related genes.
Credit: Adapted from "Epigenetics and Gene Expression," by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates
Scientists have found a unique vulnerability in some rhabdomyosarcoma tumors that could potentially be targeted with an experimental drug. Rhabdomyosarcoma is a type of cancer that forms in muscle tissue. Although it can be diagnosed at any age, it primarily affects children.
In some people with rhabdomyosarcoma, two proteins become fused together, creating an abnormal protein called PAX3-FOXO1 that drives cancer cell growth.
Rhabdomyosarcoma tumors with this fusion protein grow and spread quicker than tumors without it. But, due to the fusion protein’s relatively flat shape, it’s been hard for scientists to find drugs that block it.
In an NCI-funded study, Jun Yang, M.D., Ph.D., of St. Jude Children’s Research Hospital, and his colleagues tried a different approach: targeting an accomplice of the fusion protein. They found that the fusion protein relies on an enzyme called KDM4B to drive rhabdomyosarcoma growth and survival.
So, they turned to a recently developed drug that blocks the activity of KDM4B. When combined with chemotherapy, the KDM4B-blocking drug nearly eliminated rhabdomyosarcoma tumors in mice, according to findings published July 13 in Science Translational Medicine.
“What’s powerful about this rigorous study is that it validates KDM4 as a dependency for fusion-positive rhabdomyosarcoma,” said Marielle Yohe, M.D., Ph.D., of NCI’s Center for Cancer Research, who was not involved in the study.
But before this treatment is tested in children with rhabdomyosarcoma, “I would want to see a little bit more information,” Dr. Yohe said.
Epigenetic enzymes in childhood cancer
Every year, 350 children in the United States are diagnosed with rhabdomyosarcoma. Around one-third of those children have fusion-positive tumors, Dr. Yohe noted.
Although the standard treatments (surgery, chemotherapy, and radiation) cure most children with fusion-negative rhabdomyosarcoma, they don’t offer much hope to those with fusion-positive tumors, Dr. Yang said. Fusion-positive rhabdomyosarcoma is very aggressive and often spreads through the body.
Even for children with fusion-positive rhabdomyosarcoma who do survive, he added, the treatments can have harsh long-term side effects such as hearing loss, heart problems, and secondary cancers.
“So, there is a big challenge remaining, even for survivors,” he said.
Dr. Yang’s lab studies epigenetics, chemical changes that shift how DNA is folded and packed into the cell’s nucleus thereby controlling how genes are activated. More specifically, the team studies how epigenetic changes interact with genetic changes to accelerate cancer development and growth.
Earlier studies led by NCI researchers showed that blocking epigenetic enzymes changed the core circuitry of rhabdomyosarcoma, Dr. Yohe said. “That [approach has] since been applied to a lot of other pediatric cancers: neuroblastoma, Ewing sarcoma, and infant leukemia,” she added.
Some studies have found that the fusion proteins in these cancers rely on a family of enzymes known as KDM. KDM enzymes remove chemical marks from histones, proteins that DNA winds around. These epigenetic marks affect how tightly DNA is wound and folded.
With that in mind, Dr. Yang and his colleagues explored the role of KDM enzymes in fusion-positive rhabdomyosarcoma.
Fusion-positive rhabdomyosarcoma needs KDM4B
First, the researchers looked at the levels of various KDM enzymes in rhabdomyosarcoma tumors.
They found higher levels of KDM4B in tumors than in healthy muscle tissue of patients with fusion-positive tumors and of mice engineered to develop fusion-positive rhabdomyosarcoma.
In mice, rhabdomyosarcoma tumors genetically engineered to lack KDM4B activity grew slower than rhabdomyosarcoma tumors with normal KDM4B activity.
And in cells grown in lab dishes, a drug that blocks the activity of KDM4 enzymes, including KDM4B, killed fusion-positive rhabdomyosarcoma cells but not healthy muscle cells. KDM4 enzymes are a subgroup of the KDM family.
In mice implanted with human rhabdomyosarcoma tumors, the KDM4-blocking drug slowed tumor growth and prolonged the lives of the mice compared with a placebo. Plus, the mice didn’t lose any weight, suggesting that the drug didn’t cause serious side effects.
In combination with standard chemotherapy
The scientists also tested the KDM4-blocking drug alongside a chemotherapy pair—vincristine plus irinotecan (Camptosar)—that is a common treatment for children with rhabdomyosarcoma that has come back after initial therapy.
Separately, both treatments—the chemotherapy pair and the KDM4-blocking drug—slowed tumor growth. But together, the three-drug combination completely eliminated tumors in nearly all of the mice.
“I would have loved to have seen more studies establishing the mechanism of why the [KDM4 -blocking drug] works so well with the chemo,” Dr. Yohe said. “It's important to know why it is that the drugs work well together so that you can best take advantage of that,” she added.
An ongoing trial for children with rhabdomyosarcoma is exploring a similar approach, Dr. Yohe noted. The trial is testing chemotherapy in combination with mocetinostat, a drug that blocks enzymes that remove different epigenetic marks from histone proteins.
Opening and closing DNA
So, how do KDM4B and the PAX3-FOXO1 fusion protein work together to drive rhabdomyosarcoma growth and survival?
PAX3-FOXO1 is a transcription factor, which means that it turns specific genes on, while KDM4B makes epigenetic changes that unfold tightly packed DNA.
By unfolding DNA, KDM4B helps the fusion protein reach and turn on multiple cancer-related genes in rhabdomyosarcoma cells, the researchers found. But the KDM4-blocking drug stops KDM4B from unfolding DNA, thereby preventing the fusion protein from turning on these cancer-related genes.
Without these cancer-related genes, rhabdomyosarcoma cells can’t survive, the scientists explained.
KDM4-blocking drugs
The KDM4-blocking drug used in the study, called QC6352, is the best such drug available so far, Dr. Yang said.
“Most [KDM4] inhibitors are not potent at all. This one is very potent and selective,” meaning it blocks only the intended enzymes and not other proteins, he said.
A decade ago, there were no drugs targeting KDM4 enzymes, Dr. Yohe said. “So, just having an effective inhibitor that's available as a pill is a big deal.”
A similar drug called QC8222 is currently being tested in a clinical trial of people with advanced colon cancer, Dr. Yang added.
But before advancing QC6352 to a clinical trial for children with rhabdomyosarcoma, it would be helpful to have more information, Dr. Yohe said, such as how long the tumor shrinkage lasts and whether the drug works in other models of rhabdomyosarcoma.
Dr. Yang and his colleagues are already trying to learn more about this potential treatment approach in several ongoing lab studies, including whether QC6352 plus chemotherapy can block the metastatic spread of fusion-positive rhabdomyosarcoma in mice.